Tadalafil gehört zur Gruppe der PDE5-Hemmer und wirkt über eine hochselektive Blockade des Enzyms Phosphodiesterase Typ 5. Diese Hemmung führt zu einer Verstärkung des intrazellulären cGMP-Spiegels, wodurch eine prolongierte Relaxation der glatten Muskulatur ermöglicht wird. Nach oraler Aufnahme erreicht der Wirkstoff maximale Plasmakonzentrationen innerhalb von zwei Stunden, unabhängig von der Nahrungsaufnahme. Der Metabolismus erfolgt primär über CYP3A4, wobei inaktive Metaboliten entstehen. Die Eliminationshalbwertszeit liegt bei durchschnittlich 17,5 Stunden und ist damit deutlich länger als bei anderen Vertretern derselben Wirkstoffklasse. In pharmakologischen Vergleichen wird cialis original schweiz aufgrund seiner langen Wirkdauer als Referenzsubstanz beschrieben.
No job name
J. Phys. Chem. A 2003, 107, 5342-5351 Binary Phases of Aliphatic N-Oxides and Water: Force Field Development and Molecular Dynamics Simulation Kristine M. Kast,†,‡ Ju
1 rgen Brickmann,† Stefan M. Kast,*,†,§ and R. Stephen Berry§ Physikalische Chemie I, Technische UniVersita¨t Darmstadt, Petersenstrasse 20, 64287 Darmstadt, Germany,and Department of Chemistry, The UniVersity of Chicago, 5735 South Ellis AVenue, Chicago, Illinois 60637ReceiVed: October 29, 2002; In Final Form: April 23, 2003
Aliphatic N-oxides as cosolvents with water play an important role in stabilizing and destabilizing the structureof biopolymers such as cellulose and proteins. To allow for detailed microscopic investigations, an empiricalforce field to be used in molecular simulations is developed for two N-oxide species, N,N,N-trimethylamine-N-oxide (TMAO) and N-methylmorpholine-N-oxide (NMMO). The intra- and intermolecular force field isparametrized mainly on the basis of quantum-chemical calculations and is tested against available experimentalspectroscopic, crystallographic, and liquid state data. Special emphasis is put on the identification of transferablepotential terms in order to guide future parametrization of other species. By construction, the force field iscompatible with widely used potential functions for proteins and carbohydrates. With the resulting parameterset, molecular dynamics simulations are carried out on binary mixtures of water and N-oxides, revealingstructural features and the influence of intramolecular N-oxide flexibility. Limitations and possible extensionsof the presented models are also discussed. I. Introduction
TMAO-water interaction potential comprises a modified Cou-lomb term and a r-10/r-4/r-2 expression covering dispersion
The unique properties of solvent mixtures as compared to
and repulsion where r means the site-site distance and has not
pure phases play an increasingly important role for industrial
been tested with respect to its performance for reproducing
applications, e.g., for tuning solubility or reactivity by introduc-ing the mole fraction as an additional control variable. Further-
condensed phase experimental data. The force field was then
more, the biochemical relevance of dissolved compounds in
used in MD simulations of a single TMAO molecule in water.20
water is an important aspect of current research on protein
Zou et al. applied the force field with some adjustments in
stability and biomolecular recognition. Tertiary aliphatic N-
simulations at finite TMAO concentrations.19 They found some
oxides are remarkable species in these respects: Some are
evidence regarding changes of water-water structure and
known as good cosolvents with water for dissolving cellulose
dynamics due to the N-oxide presence and related this result to
fibers,1-6 increasing the reactivity of the swollen cellulose
the protein stabilization effect. Besides this early TMAO force
material for further derivatization in pollution-free industrial
field, quite recently a model potential function for studying
fiber processing. For instance, N-methylmorpholine-N-oxide
intramolecular H-bond dynamics in picolinic acid N-oxide has
(NMMO) in water dissolves cellulose, whereas N,N,N-tri-
been constructed and applied to the computational treatment of
methylamine-N-oxide (TMAO) does not.7 On the other hand,
TMAO abounds in marine organisms as an osmolyte counter-
In this work, a force field for two prototypical N-oxides,
acting protein denaturation provoked by urea and related osmotic
TMAO and NMMO, is developed and tested for its capability
water stress8,9 or by high-pressure conditions.10 TMAO even
to reproduce experimental data. It is intended to be balanced in
appears to play a role in possible therapies for Alzheimer’s
the sense of satisfying several requirements: (1) The functional
disease.11 Explanations on the molecular level for these phe-
form and the parameters should be compatible with common
nomena are only beginning to surface.12-19
water models and biopolymer force fields such as CHARMM23-25
To investigate into the molecular mechanism of these effects
for proteins and extensions for carbohydrates;26 (2) the force
by computational methods such as molecular dynamics (MD)
field should be as simple as possible to avoid overly expensive
simulation techniques, a force field for mixed solvents composed
computations for simulating the solvent, yet account for
of water and N-oxide species is required that is also compatible
intramolecular flexibility; (3) certain terms in the force field
with available biopolymer potential energy functions. Noto et
that represent topologically similar units should be attributed
al.20 were the first who constructed a force field for a rigid
identical, i.e., transferable parameters guiding future parametri-
TMAO model in the presence of an aqueous environment based
zation of other N-oxide species; (4) it should be applicable for
on quantum-chemical calculations of TMAO and a single water
a range of different situations such as various concentrations.
molecule within the Hartree-Fock (HF) approximation. The
Because experimental information about N-oxide systems is verylimited, the parametrization relies mainly on quantum-chemical
* To whom correspondence should be addressed. E-mail: kast@
calculations. The adequate approximations, like basis set and
pc.chemie.tu-darmstadt.de. Phone: +49 6151 165397. Fax: +49 6151164298.
inclusion of electron correlation, have been outlined in the past
† Technische Universita¨t Darmstadt.
by some of us for a number of different N-oxide/water systems.27
‡ Present address: T-Systems GEI GmbH, Goebelstr. 1-3, 64293
Although several parameters can be directly deduced from these
resources, the parametrization of a solvent force field containing
Binary Phases of Aliphatic N-Oxides and Water
J. Phys. Chem. A, Vol. 107, No. 27, 2003 5343
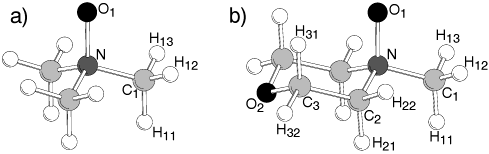
flexible molecular entities constitutes a major challenge. In thenext sections, we first describe the model function and thestrategy toward useful parameters along with the results. Themodel is validated by comparison with crystallographic andspectroscopic as well as liquid state data from experiments andis finally applied to equimolar N-oxide/water mixtures, revealingliquid-phase structural features and the influence of intramo-lecular flexibility. Figure 1. Structure and site indices for TMAO (a) and NMMO (b). II. Force Field Parametrization
structure and energetics of N-oxide hydrates from ab initio andexperimental crystal data. Second, the torsional potentials were
(a) Outline. Force field development, particularly for complex
fitted to quantum-chemical barriers, and finally, the remaining
condensed phase systems, is a challenging task, often guided
valence force field terms were determined by adjusting to
more by experience in conducting the appropriate steps than
structural and vibrational results again from ab initio calcula-
by straightforward recipes; for recent reviews, see refs 28-30
tions. The final potential was then tested against experimental
The model potential used in this work has the form
condensed phase properties by MD simulations. The force fieldis largely inspired by the CHARMM approach,23-25 in particular
we use, at least as initial estimates, known parameters from the
CHARMM force field whenever possible. By construction of
the consecutive parametrization steps, the nonbonded parameters
influence the intramolecular ones and not vice versa, so some
(r - rac)2 + ∑ ∑kabcd[1 + cos(nτ - τabcd)] +
compatibility to other force fields based on pairwise site-site
intermolecular interactions can be expected. Parameters that turn
out to be very similar upon individual optimization of similar
molecular groups will be set equal if possible without significantloss of accuracy, thereby allowing for the identification of basic
where the first three terms define an intramolecular valence force
building blocks to be used in other N-oxides. The key develop-
field and the last term the nonbonded contributions, including
ments involve the region around the N-O bond that is difficult
intermolecular interactions. Superscripts a-d denote atomic
types by requirements of topological equivalence, and subscripts
Quantum chemical ab initio calculations were performed with
i-l refer to particular atomic site indices. The intramolecular
the Gaussian suite of programs.33 The appropriate level of theory
valence force field consists of harmonic terms for bond
has been analyzed in depth in an earlier study:27 Although the
stretching (site distance rij, force constant kr, and equilibrium
HF approximation with the 6-31G** basis set is suitable for
distance r0) and angle bending (bend angle R, force constant
pure compounds, water complex properties need computations
kR, and equilibrium angle R0), and a torsional potential defined
on the MP2 level (Møller-Plesset perturbation theory to second
over cosines of the dihedral angle τ (multiplicity n, phase τ0,
order) for correctly representing experimental H-bond energies.
and torsional parameter kτ,n that is just half the energy barrier). N-oxide/water complex interactions energies were corrected by
For fine-tuning the normal frequencies, additional Urey-Bradley
the basis set superposition error (BSSE) according to Boys and
terms are introduced comprising a harmonic potential along the
Bernadi.34 MD simulations were conducted in the isothermal-
distance between the first and the third atom of a bend angle.
isobaric (NpT) ensemble35,36 at a pressure of 1 bar and various
The nonbonded potential is described basically by the sum of
temperatures, using a time step of 1 fs and periodic boundary
Coulomb interaction (nonpolarizable partial site charges q,
conditions throughout and applying distance constraints when
dielectric constant 0) and a Lennard-Jones term (well depth ,
necessary.37,38 The numerical parametrization work was done
contact distance σ; standard Lorentz-Berthelot combinations
for a large part with a dynamical simulated annealing optimiza-
ab ) ( a b)1/2, σab ) (σa + σb)/2)
(b) Partial Charges. Based on results of earlier ab initio
investigations,27 atomic site charges were determined by fitting
4 ab[(σab)12 - (σab)6]
to the electrostatic potential (ESP charges) rather than frompopulation analysis. The latter (see also ref 20) yields quite
for all atoms pairs in different molecules as well as in the same
unphysical values particularly for the central N atom that carries
molecule if they are separated by three or more bonds.
a positive formal charge. We used gas phase results instead of,
Throughout, nonbonded interactions except for intramolecular
for instance, quantum-chemical reaction field techniques to allow
distances are modified by multiplication with23,32 (1 - (rij/rc)2)2
for electronic polarization due to the environment in order to
using a truncation distance rc discussed later. For reasons of
maintain compatibility with the parametrization strategy com-
computational performance, this form has been used for both
monly used for solute species. Furthermore, for such a nonpo-
the Lennard-Jones and the Coulomb term; the energetic differ-
larizable force field as used in this work, we would need
ence as compared to applying more elaborate and computa-
representative environment models for a broad range of molar
tionally more demanding Lennard-Jones shifting techniques32
ratios between solvent and cosolvent. To allow for rotatable
is negligibly small for such strongly polar systems.
methyl groups the hydrogen charges were averaged, also for
An appropriate strategy for finding suitable parameters for
the NMMO methylene groups. Table 1 shows the dipole
the model compounds TMAO and NMMO (the structure and
moments for both the HF and the MP2 level of theory together
site numbering is shown in Figure 1) consists of several
with result from the respective point charge distribution, and
consecutive stages: First, the site charges were determined from
experimental values40,41 for TMAO. As can be seen, dipole
quantum-chemical ab initio calculations of isolated N-oxides;
moments taken directly from the wave function are quite similar
the remaining nonbonded parameters were adjusted to represent
for HF and MP2, and the HF result is closer to the experimental
5344 J. Phys. Chem. A, Vol. 107, No. 27, 2003 TABLE 1: Dipole Moments µ Resulting from Wave Function and Point Charge Distributions on Various Levels of Theory TABLE 2: Assignment of Atom Types and Partial Charges to the N-Oxidesa Figure 2. Superposition of optimized ab initio and force field structures of dihydrates of TMAO (a) and NMMO (b). TABLE 3: Lennard-Jones Parameters of the Atom Types
calculations are 0.045 Å for TMAO and 0.056 Å for NMMO
a Subscripts a-c denote atoms in topologically equivalent methyl/
with the final Lennard-Jones parameters summarized in Table
3. The BSSE-corrected quantum-chemical and resulting forcefield interaction energies (computed on the ab initio structures)
values for TMAO. However, the HF point charge result deviates
are for TMAO dihydrate -20.04 and -19.57 kcal mol-1,
strongly in the TMAO case. Therefore, the HF-derived point
respectively, and for NMMO dihydrate -18.70 and -19.31 kcal
charges were used for NMMO only and the MP2 ones for
mol-1. A number of other mono- and dihydrate structures have
TMAO. The final charges are given in Table 2 along with the
been computed yielding a rms deviation between BSSE-
assigned atom type symbols used later.
corrected MP2 energies and force field values of around 0.9
With the resulting partial charges, the potential truncation
kcal mol-1, so no further parameter adjustment was deemed
distance was optimized using a procedure developed by Dufner
et al.:42 Forces and energies from direct summation of the
It turned out that the force field dihydrate structures tend to
shifted-force potential were compared with Madelung values
break Cs symmetry by bending the water planes synchronously
from Ewald summation43 in the case of the experimental TMAO
toward the methyl/methylene groups plane upon geometry
crystal structure.44 For a cutoff distance, rc, of 13 Å, directly
optimization, regardless of the parameters. Because this effect
summed energies deviate by less than 2% and forces less than
was not observed for the ab initio structures, we can attribute it
1% from the true Madelung values. This truncation distance
to a lack of flexibility, i.e., the nonpolarizability, of the model
has then been used throughout. From earlier experiences with
potential, expected to be large at the N-oxide oxygen. Improve-
cutoff distances optimized in this way,39 we can expect structural
ment could possibly be achieved by using off-site charge centers
deviations with respect to results from crystal simulations
on the oxygen atom, reflecting to some extent charge transfer
applying the Ewald summation technique of around 1-2%.
into the water hydrogen directions. Both possible remedies,
(c) Lennard-Jones Parameters. Because no explicit polar-
adding explicit polarizability49,50 or off-site charge centers for
izability was taken into account, the remaining nonbonded
flexible entities, would mean a significant complication of the
parameters were designed to reflect the effective many-body
model and higher computational demand. We therefore refrained
interactions mapped on simple site-site interactions. The
from such extensions and used constraints for keeping the water
Lennard-Jones parameters in the N-O region were adjusted to
molecules upright during geometry optimizations, mimicking
geometrical and energetic reference data from ab initio calcula-
the directional forces induced by the H-bonding environment
tions on N-oxide hydrates. The dominant structural motif found
in the experimental crystal structures can be reproduced by
The ether group in the morpholine ring (-CH -
geometry optimization of TMAO and NMMO dihydrates on
the Lennard-Jones parameters of which were set to standard
the MP2 level (see Figure 2). In this case, the internal N-oxide
values of the CHARMM parameter set up to this point, needed
geometries were frozen on the previous results,27 and the water
some further refinement: The associated parameters were
structure and potential was represented by the rigid three-site
slightly modified by a Newton-Raphson51 minimization of a
TIP3P model45,46 with small Lennard-Jones parameters attributed
to the hydrogens.47 The parameters of the atoms in the N-Oregion including attached methyl and methylene groups were
S( ) ) ∑(〈O 〉 - O )2
then adjusted by simulated annealing28,39 in order to reproduce
the optimal dihydrate geometries. Each atom sort was assignedthe same parameters, taken to be transferable between TMAO
that measures the deviation of thermally averaged observables
Oi( ) depending on a number of parameters
As a result, illustrated in Figure 2, rms deviations between
values Oi,ref. In our case, the crystallographic cell parameters
matched48 dihydrate structures from ab initio and force field
of NMMO monohydrate act as observables, with reference
Binary Phases of Aliphatic N-Oxides and Water
J. Phys. Chem. A, Vol. 107, No. 27, 2003 5345 TABLE 4: Torsional Parameters kabcd a TABLE 5: Rigid Substructure Coordinates for the Partly Rigid TMAO Model TABLE 6: Rigid Substructure Coordinates for the Partly Rigid NMMO Model a Multiplicity n ≡ 3, phase τ
0 ≡ 0; a-d denote adjacent atoms
values taken from the literature52 and thermal averages obtained
from MD simulations with the rigid molecules used up to this
point. Also, the necessary first and second derivatives of S with
respect to nonbonded parameters were approximated by finite
differences, deduced from a number of 48 ps simulations at
298.15 K and 1 bar with small variations of the Lennard-Jones
parameters applied to a crystal section comprising 384 water
and NMMO molecules. It turned out that the cell parameters
model to be examined and contrasted with the fully flexible
are only weakly sensitive to a variation of the ether group
model in more detail later. We do not consider the inherently
nonbonded parameters, so the optimization was terminated after
quantal nature of methyl group rotations in our model. The rigid
a single Newton-Raphson step. The resulting parameters are
substructure coordinates are given in Tables 5 and 6; the
remaining constraint distances are d(N-H
(d) Torsional Parameters. Starting from HF-optimized N-oxide geometries with imposed local C
methyl groups, the ab initio torsional energy profile was
11-H12) ) d(H12-H13) ) d(H11-H13) ) 1.772
computed on the MP2 level by varying the O-N-C-H dihedralangle in steps of 30°, keeping all other coordinates fixed. The
(e) Bond and Angle Parameters. Keeping all parameters
corresponding force field barriers, to be expressed by the
determined so far fixed, the remaining bond stretching and angle
torsional potential terms, were then computed from the differ-
bending terms were adjusted with respect to minimizing the
ence of ab initio energies and the model potential known so
deviations between quantum-chemical and force field N-oxide
far. To this end, inclusion of 1-4 Coulomb plus Lennard-Jones
optimal structures and normal vibrations. Using again HF/6-
as well as only 1-4 Lennard-Jones interactions, both without
31G** N-oxide geometries, the normal frequencies were
further scaling, were compared. The energy barriers obtained
determined and scaled by the empirical value of 0.893.53 The
are very similar for TMAO and NMMO in both cases, including
force field parameters were optimized again by simulated
only 1-4 Lennard-Jones terms (TMAO 0.540 kcal mol-1,
annealing;28,39 structural data in the form of atomic distances
NMMO 0.537 kcal mol-1) or including both Lennard-Jones and
and vibrational data were weighted equally, and normal modes
Coulomb interactions (TMAO 0.506 kcal mol-1, NMMO 0.506
were assigned by maximizing the overlap between model and
kcal mol-1). To allow for transferability to other N-oxides with
different charge distributions, torsional parameters obtained from
Starting with TMAO, it turned out that the inclusion of Urey-
the calculations including only 1-4 Lennard-Jones contributions
Bradley terms is essential for a reliable representation of
were derived for the further parametrization process.
structure and normal mode spectrum. With the resulting
Assuming equal contributions to the total barrier, the indi-
parameter set given in Tables 7 and 8, the rms deviation between
vidual terms were distributed (for instance one H-C-N-C and
matched force field and ab initio geometry is 0.005 Å, the rms
two H-C-N-O terms for each of three hydrogen atoms in
frequency deviation is ca. 50 cm-1. The spectrum is depicted
one methyl group, the result multiplied by three gives the total
in Figure 3 together with experimental infrared-spectroscopic
barrier) and averaged for TMAO and NMMO. Force field
data by Kuroda and Kimura.54 More detailed frequency infor-
contributions to the dihedral potential within the NMMO
mation along with other experimental sources55,56 is summarized
morpholine ring were described by standard CHARMM and
in Table 9, showing excellent agreement. The normal mode
carbohydrate26 parameters for similar torsions. In this way, the
quality can be directly attributed to the global optimization
methyl group rotation is correctly described in all cases, whereas
technique applied, and any simple local optimization with
the torsion contribution to the intraring flexibility of NMMO
starting values taken from similar fragments fails.
influences the normal vibrations that are optimized in the next
Keeping parameter transferability to other N-oxides in mind,
section by adjusting the remaining parameters. All torsional
as many TMAO bond and angle terms as possible were used
without change for the N-O region in NMMO. Furthermore,
The set of intermolecular and torsional potential terms
the intraring torsional potentials were not modified in anticipa-
allowing only for methyl group rotation constitutes a partly rigid
tion that the spectral adjustment could be accomplished solely
5346 J. Phys. Chem. A, Vol. 107, No. 27, 2003 TABLE 7: Harmonic Bond Stretching Parameters: Force Constants k ab ab and Equilibrium Distances r0
by varying the morpholine bend angle potentials. For correctlyreproducing the angle between the N-O axis and the ring, itturned out that the O -
released, thereby dropping some transferability of the intramo-lecular parameters of the N-O region. This is related to thestrong electrostatic oxygen-oxygen repulsion that could againin principle be compensated by off-site oxygen charge centersor explicit polarizability. Transferable values were maintainedfor the parameters of the O -
bond potentials as well as for the parameters of the N-C -
angle potential and the corresponding N-H1 Urey-Bradleyterm. New NMMO parameters had to be derived for the O -
N-C1 angle potential and for all bond and angle potentials thatimply other morpholine atoms besides nitrogen. Additionally,Urey-Bradley distance potentials for all 1-3 atom pairs exceptfor O -
C2 and N-C3 were necessary. With the final parameter
set given in Tables 7 and 8, the structural rms deviation between
Figure 3. Scaled ab initio and force field normal frequencies of TMAO (top, with experimental IR spectrum54) and NMMO (bottom). Dashed
force field and ab initio results is 0.004 Å, and the rms frequency
deviation is ca. 48 cm-1. The NMMO spectrum is also depictedin Figure 3, indicating the excellent quality of the intramolecular
optimized N-oxide geometry with local C3 methyl symmetry,
where only the methyl groups are free to rotate under the actionof the torsional potential, and the fully flexible model using all
III. Molecular Dynamics Simulation Results
The complete force field was tested by MD simulations of
(a) Crystal Structures. Unit cells of NMMO,52 NMMO
condensed phases of N-oxide/water systems, both crystalline
monohydrate,52 di-NMMO-pentahydrate,57 and TMAO dihy-
and liquid ones, at various conditions for which experimental
drate58 were multiplied to yield reasonable simulation boxes (a,
data are available. Two model instances were taken into
b, c multiples were for NMMO: 4 × 5 × 7, NMMO‚H2O: 2
account: The partly rigid model based on the HF/6-31G**-
× 6 × 4, 2NMMO‚5H2O: 3 × 6 × 2, TMAO‚2H2O: 4 × 3 ×
TABLE 8: Harmonic Angle Bending and Associated Urey-Bradley 1-3 Stretching Parameters: Force Constants kabc ac ac a
R /k and Equilibrium Displacements R /r a a-c denote adjacent atoms constituting a bend angle.
Binary Phases of Aliphatic N-Oxides and Water
J. Phys. Chem. A, Vol. 107, No. 27, 2003 5347 TABLE 9: Vibrational Frequencies of TMAO: Computed
density deviation of more than 20%. Given the quality of agree-
(HF/6-31G**, FF: Force Field) and Experimental: (a)
ment for all other phases studied, one might speculate that the
Gigue`re and Chin,55 (b) Kuroda and Kimura,54 and (c)
experimental crystal structure is flawed: The experimental struc-
Choplin and Kaufmann56
ture consists of antiparallel TMAO layers perpendicular to the
a axis; the smallest site-site distance observed between the
layers is 2.951 Å (hydrogen-hydrogen) while within the layers
the shortest distance is 1.615 Å (also hydrogen-hydrogen). Such
an anomalously large gap does not exist in the other densely
packed N-oxide and N-oxide hydrate structures. (b) Density of Liquid Mixtures. A number of simulation
for various molar ratios of N-oxide/water and two different
temperatures have been carried out from which the average
densities were obtained. These could be compared with experi-
mental values.7 For the 1:5 N-oxide/water ratio, a total of 840
molecules were used, 1034 for the 1:10 mixture, and 1120 for
1:15. The TMAO 1:10 system corresponds roughly to a 4 M,
and the TMAO 1:15 system corresponds to a 3 M solution.19
Starting with randomly placed molecules, the systems were
equilibrated for 400 ps at 1000 K and constant volume at the
expected density, and for another 40 ps at the specified
temperature and 1 bar followed by 100 ps NpT sampling runs.
The results are summarized in Table 11.
The agreement between experimental and computational
results is generally good, and even better so for NMMO. TMAO
solutions tend to be systematically denser than obtained
experimentally, whereas NMMO solutions are less dense,
TABLE 10: Results of NpT Simulations and Experimental
although to a lesser percentage. Accounting for full flexibility
Crystal Data of NMMO and NMMO Monohydrate (exp., ref
shows an albeit small yet notably systematic effect for the
52), Di-NMMO Pentahydrate (exp., ref 57), and TMAO
solutions: The densities in general slightly increase. The smaller
Dihydrate (exp., ref 58) at 293.15K and 1 Bara
the N-oxide concentration, the smaller the density deviations
as expected, because for small molarity the properties of the
water model dominate that is optimized for bulk properties.
Further optimization of the N-oxide models should focus on
the N-O oxygen polarization: As we have seen during the
parametrization, with the present point charge model, water
molecules do not keep the correct orientation relative to the
N-O group. This feature is most likely responsible for the slight
(c) Liquid Structure of Equimolar Mixtures. We finally
turned to conditions for which no experimental information is
available but that are most important for the problem of cellulose
solubility: Equimolar mixtures of NMMO and water do dissolve
cellulose, whereas analogous TMAO solutions do not.7 We
cannot expect to explain these phenomena from structural
properties of the solvent alone, but the results will serve as a
reference for characterizing the influence of a solute to be
studied in the future. Simulation systems were prepared
a Cell parameters a-c, monoclinic angle , average density F.
analogously to the dilute solutions: For TMAO/water, 600molecules were used at 1 bar and a temperature of 533.15 K,
3). The systems were simulated at the experimental conditions
above the melting point of the amorphous monohydrate of
of 293.15 K and 1 bar for 100 ps after 150 ps of equilibration.
474.15 K as reported by Hattori.59 For the NMMO/water
Table 10 shows the simulations results for cell parameters and
mixtures, 440 molecules were simulated at 1 bar and 373.15
density along with the experimental values.
K, the temperature used in industrial cellulose processing, also
The agreement is good, and the differences between partly
above the experimental melting point of 345.15 K.2 The
rigid and fully flexible models are marginal. The densities are
sampling time was 300 ps for each system.
on average improved upon using the fully flexible systems and
In contrast to the dilute solutions, we observe in the equimolar
deviate on average by around 1% from the experimental values.
case a rather strong dependence on the chosen model, more
The largest discrepancies are observed for the monoclinic angle
visible for TMAO: The average density from the partly rigid
in the NMMO cases. One can again expect that the single local-
model is 0.672 g cm-3, and for the fully flexible one, it is 0.764
ized point charge on the N-oxide oxygen is responsible for this
g cm-3. For NMMO, we have 1.065 (partly rigid) and 1.093 g
effect: The true charge distribution is slightly shifted off the
cm-3 (fully flexible). Accounting for full flexibility obviously
morpholine ring, accounting for which would induce a change
increases the density (by 12% for TMAO and 2.5% for NMMO),
in the relative NMMO orientations. Simulation details of pure
as seen before for the dilute systems to a lesser extent. Inter-
TMAO are not given here. All cell parameters agree quite well
and intramolecular degrees of freedom strongly couple under
with the experimental results44 except for the a axis, yielding a
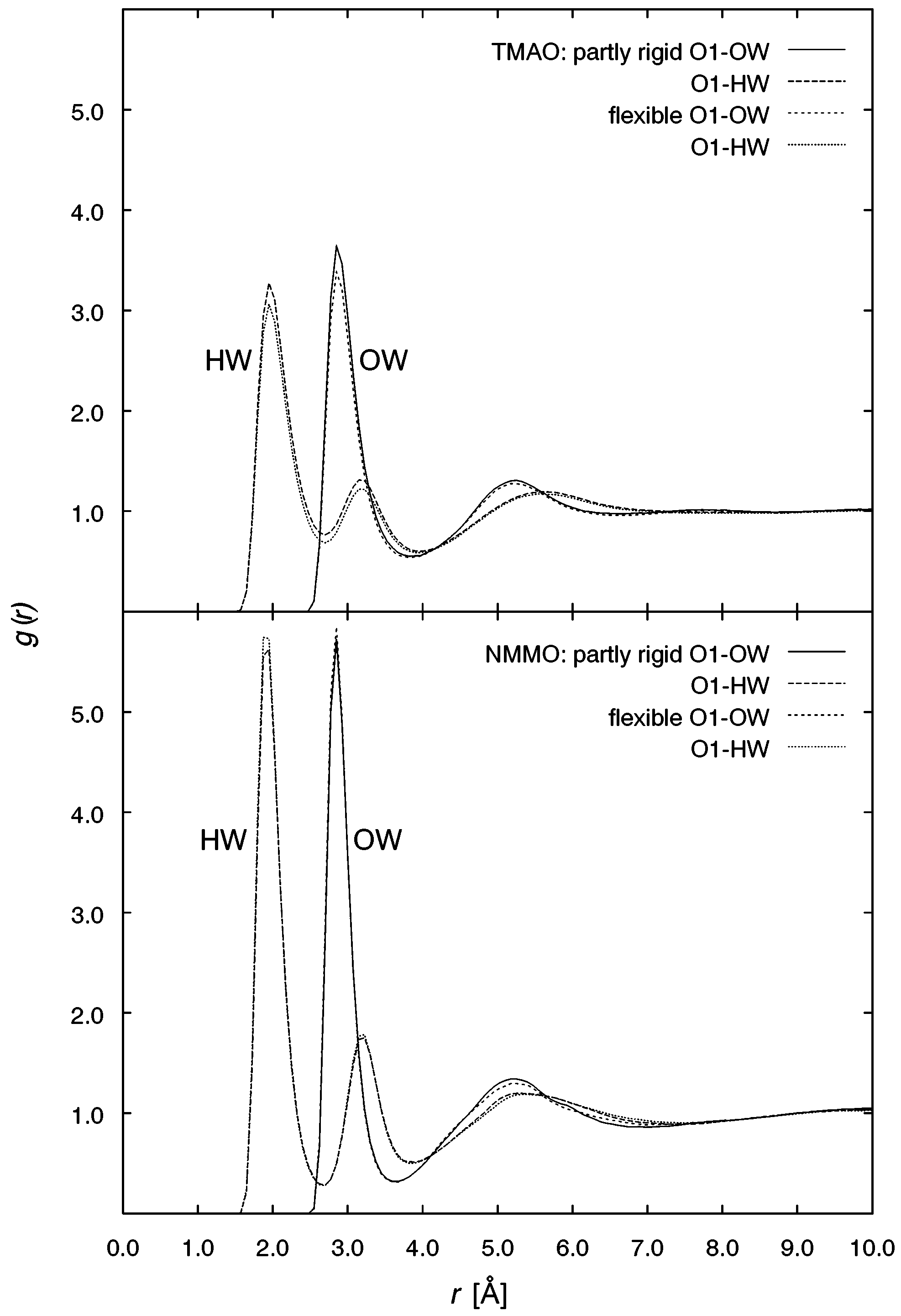
these conditions, unexpectedly larger so for TMAO. 5348 J. Phys. Chem. A, Vol. 107, No. 27, 2003 TABLE 11: Average Densities G from NpT Simulations and Experimental Data7 of Liquid Mixtures of TMAO or NMMO and Water at Various Temperatures T and Molar Ratios N-Oxide/Water Figure 4. Radial distribution functions of the water oxygen OW around Figure 5. Radial distribution functions of the water oxygen OW and
the N-oxide atoms O1, N, C1, and O2 (only for NMMO), for TMAO/
hydrogen HW around the N-oxide oxygen O1, for TMAO/water (top)
water (top) and NMMO/water (bottom).
This phenomenon should also be reflected by the liquid
O2 distance of 4.27 Å found for one of the NMMO
structure that is analyzed here in terms of radial distribution
monohydrate structures obtained from earlier ab initio calcula-
functions, g(r), for various site pairs. In the case of N-oxide/
tions27 where the water molecule is positioned above the
water mixtures, the formation of solvent shells of water around
morpholine ring bridging both NMMO oxygens. A slight
the polar N-O bond as well as the positions of N-oxides around
influence of flexibility can be observed in these g functions.
each other are of great importance. The water oxygen OW
In Figures 5 and 6 (top), the distribution of water sites around
distribution around the atoms of the functional N-oxide group
the oxygen atoms in TMAO and NMMO is shown. For the
O1, N, C1 (and for NMMO also O2 in the morpholine ring) is
oxygen atoms O1 of the N-O group (Figure 5), in both systems,
shown in Figure 4. Concerning the N-O oxygen, two solvent
two solvent shells of the water oxygen OW and three peaks for
shells at around 2.8 and 5.2 Å can be found which appear to be
the corresponding hydrogen atoms HW can be observed, again
more pronounced in the NMMO case because of the lower
more pronounced for NMMO. The corresponding H-bond
temperature. The distribution around the carbon atom C
distances for HW O1 of about 1.9 Å are in good agreement
rotatable methyl groups shows for TMAO two weak maxima
with the results of ab initio calculations of N-oxide monohy-
at around 3.2 and 5.0 Å. For NMMO, the solvation shell is
drates.27 In Figure 5, the first two peaks for HW and the first
characterized by a single predominant peak. The distribution
OW peak can be assigned to the same water molecule. As the
around the nitrogen atom appears for both systems as a broader
first HW peak is much larger than the second one, an H-bond
maximum at around 4.0 Å. For the ring oxygen O2 of NMMO
with a more mobile water molecule can be assumed. The
also two solvent shells can be identified with the second peak
position of this water molecule corresponds to the optimized
at ca. 4.8 Å as the more pronounced one. This value resembles
structure of the monohydrates for TMAO and NMMO.27
Binary Phases of Aliphatic N-Oxides and Water
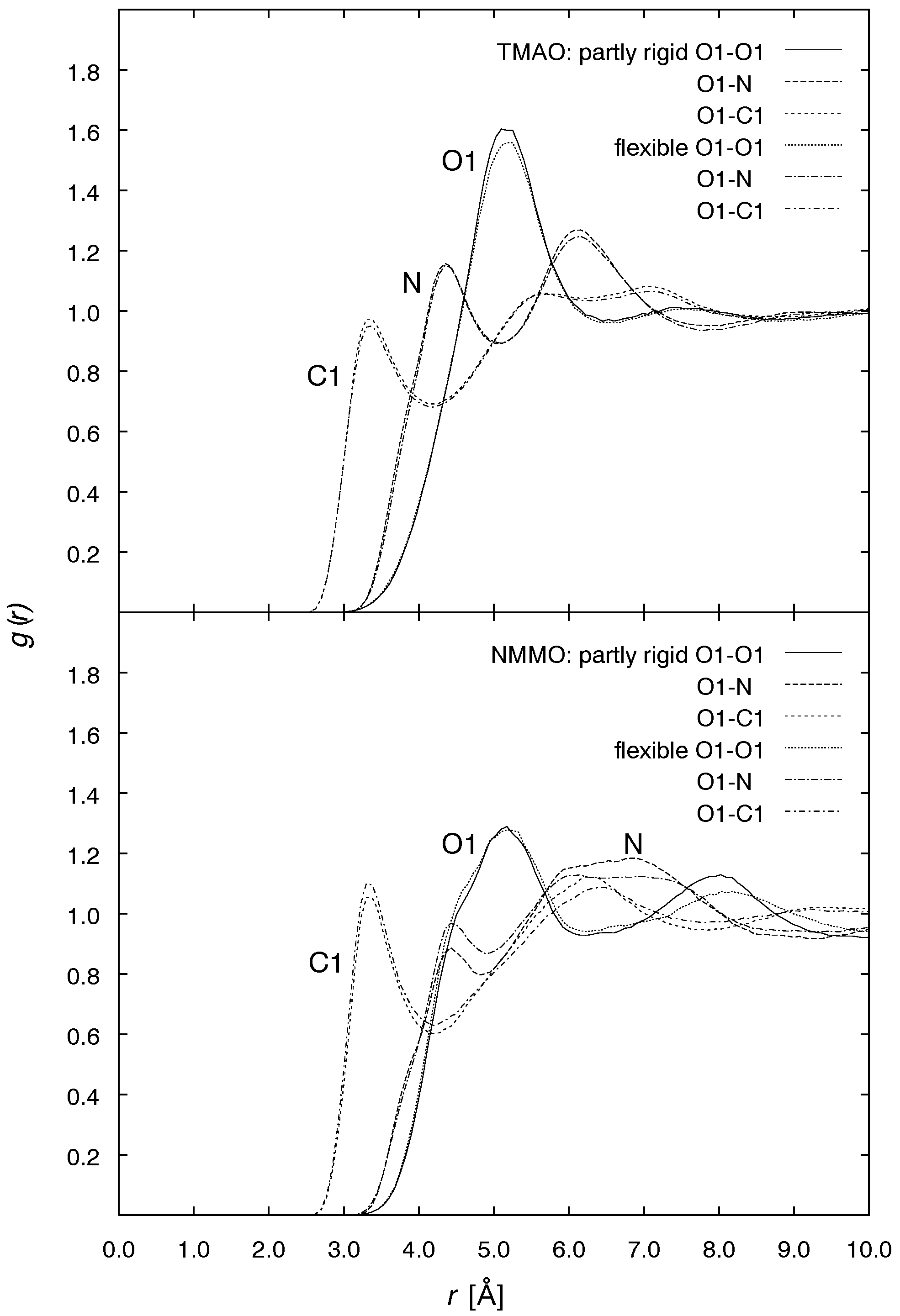
J. Phys. Chem. A, Vol. 107, No. 27, 2003 5349 Figure 6. Radial distribution functions of the water oxygen OW and Figure 7. Radial distribution functions of the N-oxide atoms O1, N,
hydrogen HW around the NMMO ring oxygen O2 (top) and of the
and C1 around the N-oxide oxygen O1, for TMAO/water (top) and
NMMO atoms O1, N, and C1 around the NMMO atom O2 (bottom).
By calculating the average amount of OW atoms in a region
Finally, in analogy to the distribution of OW (Figure 4), the
1, for each of the N-oxides, more than one
water molecule in the direct neighborhood of the N-O group
distribution of the methyl group’s atoms O1, N, and C1 around
can be found. For the partly rigid TMAO, we have 1.12 water
O1 is shown in Figure 7. Comparing the results for TMAO and
molecules, whereas the flexible model yields 1.20. For NMMO,
NMMO, both show more than one N-oxide coordinated with
the average number of surrounding water molecules increases
the O1 atom. The distribution of the NMMO molecules shows
from 1.28 to 1.32 upon switching to full flexibility. Water is
markedly more pronounced differences between the partly rigid
apparently more tightly bound to the N-O group if flexible
and the flexible model than the TMAO distribution due to the
molecules are present, more so for TMAO than for NMMO.
flexible ring system. The onset of the peaks at ca. 3 Å point to
To clarify the physical nature of this effect, we will have to
the existence of bridging water molecules between the N-oxides.
look at intra-/intermolecular cross correlation functions and
IV. Concluding Remarks
vibrational/librational mode coupling, for which much largersimulation times will be necessary.
The present paper aimed at the development and assessment
In the case of the morpholine ring oxygen atom O2 (Figure
of an empirical force field for aliphatic N-oxides as important
6, top), more water molecules can be found in the second solvent
cosolvents for water with interesting and still unexplained
shell than in the first one, and it is also spread more broadly.
properties with respect to the stabilization of biomolecules in
This is a hint for a water position directly above the morpholine
solution. We have focused on the following key issues: (i) The
ring. Compared to the N-O oxygen O1, a much smaller number
thorough derivation of potential parameters from a variety of
of water molecules can be found in the O2 region, and the peaks
sources by advanced parametrization techniques, (ii) the iden-
for the partly rigid and the flexible model show slight differ-
tification of basic building blocks guiding future parametrization
ences. This effect is even more obvious in the distribution of
of related species, (iii) providing simulation results as reference
atoms of the N-O group (O1, N, and C1) around the ring oxygen
material for studies of solvated molecules, and (iv) an assess-
O2 (Figure 6, bottom). For the carbon C1 of the rotating methyl
ment of the influence of molecular flexibility as a likely source
group, only a weak solvent shell can be observed around O2.
of genuine N-oxide/water mixture properties.
The peaks for O1 and N are similarly high and broad. For O1
The potential function derived for the two prototypical
on the other hand, there exists another solvent shell that is lower
N-oxide species TMAO and NMMO yields single molecule and
than the first one in the flexible model. The similar height of
condensed phase properties in good agreement with available
the main peaks for O1 and N and the distance between those
experimental data, given the requirements of simplicity and
peaks corresponding to the length of the N-O bond hint at an
transferability outlined in the Introduction. Because of the simple
O1 N orientation mediated by water molecules.
functional form chosen, standard parameter combination rules
5350 J. Phys. Chem. A, Vol. 107, No. 27, 2003
apply for modeling ternary phases composed of biopolymer
(3) Chanzy, H.; Noe, P.; Paillet, M.; Smith, P. J. Appl. Polym. Sci.:
species and an N-oxide/water solution, allowing for detailed
Appl. Polym. Symp. 1983, 37, 239.
(4) Taeger, E.; Michels, C.; Nechwatal, A. Das Papier 1991, 12, 784.
microscopic investigations. With the present force field, first
(5) Creely, J. J.; Wade, R. H.; French, A. D. Text. Res. J. 1978, 48,
simulations of an oligosaccharide acting as a cellulose model
in solvent mixtures of TMAO or NMMO and water have already
(6) Philipp, B.; Schleicher, H.; Wagenknecht, W. Chem. Technol. 1977,
been conducted,60 revealing certain H-bond patterns between
(7) Akzo Nobel, Obernburg, private communication.
the carbohydrate and NMMO that could explain the solubility
(8) Yancey, P. H.; Somero, G. N. J. Exp. Zool. 1980, 212, 205.
of cellulose in NMMO/water mixtures. Interestingly though, the
(9) Yancey, P. H.; Clark, M. E.; Hand, S. C.; Bowlus, R. D.; Somero,
stabilization of proteins by diluted TMAO appears to be related
G. N. Science 1982, 217, 1214.
indirectly to a change of the water structure.19 The distribution
(10) Yancey, P. H.; Siebenaller, J. F. J. Exp. Biol. 1999, 202, 3597. (11) Eidenmu¨ller, K.; Fath, T.; Hellwig, A.; Reed, J.; Sontag, E.; Brandt,
functions for binary mixtures described in this work define a
R. Biochemistry 2000, 39, 13166.
reference based on which the perturbing influence of a polymeric
(12) Anthoni, U.; Christophersen, C.; Gajhede, M.; Nielsen, P. H. Struct.
solute can be studied in the future. The remarkable effect of
Chem. 1992, 3, 121.
intramolecular N-oxide flexibility beyond methyl group rotations
(13) Lin, T. Y.; Timasheff, S. N. Biochemistry 1994, 33, 12695. (14) Wang, A.; Bolen, D. W. Biochemistry 1997, 36, 9101.
that has been observed for binary mixtures appears also to play
(15) Baskakov, I. V.; Bolen, D. W. J. Biol. Chem. 1998, 273, 4831.
a role for the solvation process, although no quantitative
(16) Baskakov, I. V.; Kumar, R.; Srinivasan, G.; Ji, Y.; Bolen, D. W.;
assessment based for instance on free energy computations has
Thompson, E. B. J. Biol. Chem. 1999, 274, 10693.
been attempted yet. Detailed discussions of the flexibility
(17) Palmer, H. R.; Bedford, J. J.; Leader, J. P.; Smith, R. A. J. J. Biol.Chem. 2000, 275, 27708.
problem will require much more expensive simulations than
(18) Sharp, K. A.; Madan, B.; Manas, E.; Vanderkooi, J. M. J. Chem.Phys. 2001, 114, 1791.
(19) Zou, Q.; Bennion, B. J.; Daggett, V.; Murphy, K. P. J. Am. Chem.
The final parameter set serves as good starting point for the
Soc. 2002, 124, 1192.
parametrization of other N-oxides: Keeping various terms
(20) Noto, R.; Martorana, V.; Emanuele, A.; Fornili, S. L. J. Chem.
unchanged that have been identified as being transferable, future
Soc., Faraday Trans. 1995, 91 3803.
force field development can focus solely on unknown parts of
(21) Stare, J.; Mavri, J.; Ambrozˇicˇ, G.; Hadzˇi, D. J. Mol. Struct.(THEOCHEM) 2000, 500, 429.
the compounds under consideration. The available evidence
(22) Dosˇlic´, N.; Stare, J.; Mavri, J. Chem. Phys. 2001, 269, 59.
gathered throughout the parametrization and validation process
Brooks, B. R.; Bruccoleri, R. E.; Olafsen, B. D.; States, D. J.;
points to a key limitation of the present force field that needs
Swaminathan, S.; Karplus, M. J. Comput. Chem. 1983, 4, 187.
to be addressed in future work: The simple point charge model
(24) Nilsson, L.; Karplus, M. J. Comput. Chem. 1986, 7, 591. (25) MacKerell, A. D., Jr.; Bashford, D.; Bellott, R. L.; Dunbrack, R.
of the N-oxide oxygen appears not to be capable of describing
L., Jr.; Evanseck, J. D.; Field, M. J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.;
the physical situation satisfactorily; adding a polarization term
Joseph-McCarthy, D.; Kuchnir, L.; Kuczera, K.; Lau, F. T. K.; Mattos, C.;
will be necessary to account for certain structural features of
Michnick, S.; Ngo, T.; Nguyen, D. T.; Prodhom, B.; Reiher, W. E., III.;
pure and hydrated N-oxides. Another issue to consider is the
Roux, B.; Schlenkrich, M.; Smith, J. C.; Stote, R.; Straub, J.; Watanabe, M.; Wiorkiewicz-Kuczera, J.; Yin, D.; Karplus, M. J. Phys. Chem. B 1998,
water model that has been restricted in this work to the rigid
TIP3P form optimized for the neat liquid: One not only needs
(26) Reiling, S.; Schlenkrich, M.; Brickmann, J. J. Comput. Chem. 1996,
to address the problem of parameter validity at high cosolvent
(27) Kast, K. M.; Reiling, S.; Brickmann, J. J. Mol. Struct. (THEOCHEM)
concentration but also the intramolecular water dynamics that
1998, 453, 169.
appears to be modulated in the presence of TMAO, as indicated
(28) Kast, S. M.; Brickmann, J.; Berry, R. S. In Conceptual PerspectiVes
by a recent infrared-spectroscopic study.18 It will be interesting
in Quantum Chemistry; Calais, J. L., Kryachko, E., Eds.; Kluwer: Dordrecht,
to see what level of physical detail is necessary to explain the
(29) Norrby, P.-O.; Liljefors, T. J. Comput. Chem. 1998, 19, 1146.
genuine N-oxide effects on biopolymer stability.
(30) Wang, J.; Kollman, P. A. J. Comput. Chem. 2001, 22, 1219.
A number of open questions can only be answered by
(31) Henderson, D. Annu. ReV. Phys. Chem. 1974, 25, 461.
appropriate experiments performed on N-oxide/water mixtures.
(32) Steinbach, P. J.; Brooks, B. R. J. Comput. Chem. 1994, 15, 667. (33) Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.;
For instance, frequencies and amplitudes of isotopically sub-
Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T.; Petersson, G.
stituted species should give rise to different structures and
A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski,
thermodynamics. Equally important would be information about
V. G.; Ortiz, J. V.; Foresman, J. B.; Cioslowski, J.; Stefanov, B. B.;
the dielectric constants for various mixtures as well as studies
Nanayakkara, A.; Challacombe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.;Wong, M. W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.;
with respect to a separation of intramolecular and intermolecular
Fox, D. J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head-
correlation functions that could be directly compared with
Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian 94; Gaussian, Inc.:
simulation data. Perhaps this work can inspire some experi-
(34) Boys, S. F.; Bernadi, F. Mol. Phys. 1970, 19, 553.
(35) Kast, S. M.; Brickmann, J. J. Chem. Phys. 1996, 104, 3732. (36) Berendsen, H. J. C.; Postma, J. P. M.; van Gunsteren, W. F.; DiNola, Acknowledgment. This work was supported by the Deutsche
A.; Haak, J. R. J. Chem. Phys. 1984, 81, 3684.
(37) Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H. J. C. J. Comput. Phys.Forschungsgemeinschaft within the Schwerpunktprogramm
1977, 23, 327.
“Cellulose und Cellulosederivate - molekulares und supramole-
(38) Ciccotti, G.; Ferrario, M.; Rykaert, J.-P. Mol. Phys. 1982, 55, 549.
kulares Strukturdesign”. S.M.K. thanks the Alexander von
(39) Hauptmann, S.; Dufner, H.; Brickmann, J.; Kast, S. M.; Berry, R.
Humboldt-Stiftung for financial support. Bernd Schilling pro-
S. Phys. Chem. Chem. Phys. 2003, 5, 635.
(40) Linton, E. P. J. Am. Chem. Soc. 1940, 62, 1945.
vided the programs for computing the radial distribution
(41) Phillips, G. M.; Hunter, J. S.; Sutton, L. E. J. Chem. Soc. 1945,
(42) Dufner, H.; Kast, S. M.; Brickmann, J.; Schlenkrich, M. J. Comput.Chem. 1997, 18, 660. References and Notes
(43) Ewald, P. P. Ann. Phys. 1921, 64, 253. (44) Caron, A.; Palenik, G. J.; Goldish, E.; Donohue, J. Acta Crystallogr.
(1) Chanzy, H.; Dube´, M.; Marchessault, R. H. J. Polym. Sci.: Polym.1964, 17, 102. Lett. Ed. 1979, 17, 219.
(45) Jorgensen, W. L. J. Am. Chem. Soc. 1981, 103, 335.
Chanzy, H.; Nawrot, S.; Pe´guy, A.; Smith, P. J. Polym. Sci.:
(46) Jorgensen, W. L.; Chandrasekhar, J.; Madura, J. D.; Impey, R. W.;
Polym. Phys. Ed. 1982, 20, 1909.
Klein, M. L. J. Chem. Phys. 1983, 79, 926.
Binary Phases of Aliphatic N-Oxides and Water
J. Phys. Chem. A, Vol. 107, No. 27, 2003 5351
(47) Reiher, W. E., III. Ph.D. Thesis, Harvard University, Cambridge,
(53) Pople, J. A.; Krishnan, R.; Schlegel, H. B.; DeFrees, D.; Binkley,
J. S.; Frisch, M. J.; Whiteside, R. F.; Hout, R. F.; Hehre, W. J. Int. J.
(48) Ferro, D. R.; Hermans, J. Acta Crystallogr. 1977, A 33, 345. Quantum Chem., Quantum Chem. Symp. 1981, 15, 269.
(49) Jordan, P. C.; van Maaren, P. J.; Mavri, J.; van der Spoel, D.;
(54) Kurodo, Y.; Kimura, M. Spectrochim. Acta 1966, 22, 47.
Berendsen, H. J. C. J. Chem. Phys. 1995, 103, 2272.
(55) Gigue`re, P. A.; Chin, D. Can. J. Chem. 1961, 39, 1214.
(50) Kaminski, G. A.; Stern, H. A.; Berne, B. J.; Friesner, R. A.; Cao,
(56) Choplin, F.; Kaufmann, G. Spectrochim. Acta 1970, 26A, 2113.
Y. X.; Murphy, R. B.; Zhou, R.; Halgren, T. A. J. Comput. Chem. 2002,
(57) Maya, E.; Pe´rez, S. Acta Crystallogr. 1982, B 38, 848.
(51) Press, W. H.; Teukolsky, S. A.; Vetterling, W. T.; Flannery, B. P.
(58) Mak, T. C. W. J. Mol. Struct. 1988, 178, 169. Numerical Recipes in Fortran, 2nd ed.; Cambridge University Press:
(59) Hattori, Y. J. Pharm. Soc. Jpn. 1940, 60, 24.
(60) Marho¨fer, R. J.; Kast, K. M.; Schilling, B.; Ba¨r, H.-J.; Kast, S. M.;
(52) Maya, E.; Peguy, A.; Pe´rez, S. Acta Crystallogr. 1981, B 37, 1858.
Brickmann, J. Macromol. Chem. Phys. 2000, 201, 2003.
Science, bitches: it works Kris King – May 13th 2012 Disgracefully, I haven’t written a blog post in a little over a month; predictably, I have an excuse; surprisingly, it’s a really good one. Since easter weekend my partner, Raven, and I have been engaged in a seemingly never-ending battle against the forces of contagion present in the numerous furry creatures we’ve chosen to ta
Contact: David A. Barry Sugarman, Rogers, Barshak & Cohen, P.C. (617) 227-3030 [email protected] CANADIAN COMPANY SUED FOR DEFRAUDING THOUSANDS OF U.S. CONSUMERS OF DIETARY SUPPLEMENTS BOSTON, September 16, 2008 – Consumers in sixteen states have filed lawsuits against Canadian company Wellnx Life Sciences, which markets and sells three dietary supplements
 Binary Phases of Aliphatic N-Oxides and Water
J. Phys. Chem. A, Vol. 107, No. 27, 2003 5343
Binary Phases of Aliphatic N-Oxides and Water
J. Phys. Chem. A, Vol. 107, No. 27, 2003 5343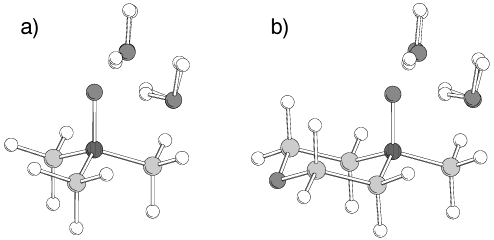 5344 J. Phys. Chem. A, Vol. 107, No. 27, 2003
5344 J. Phys. Chem. A, Vol. 107, No. 27, 2003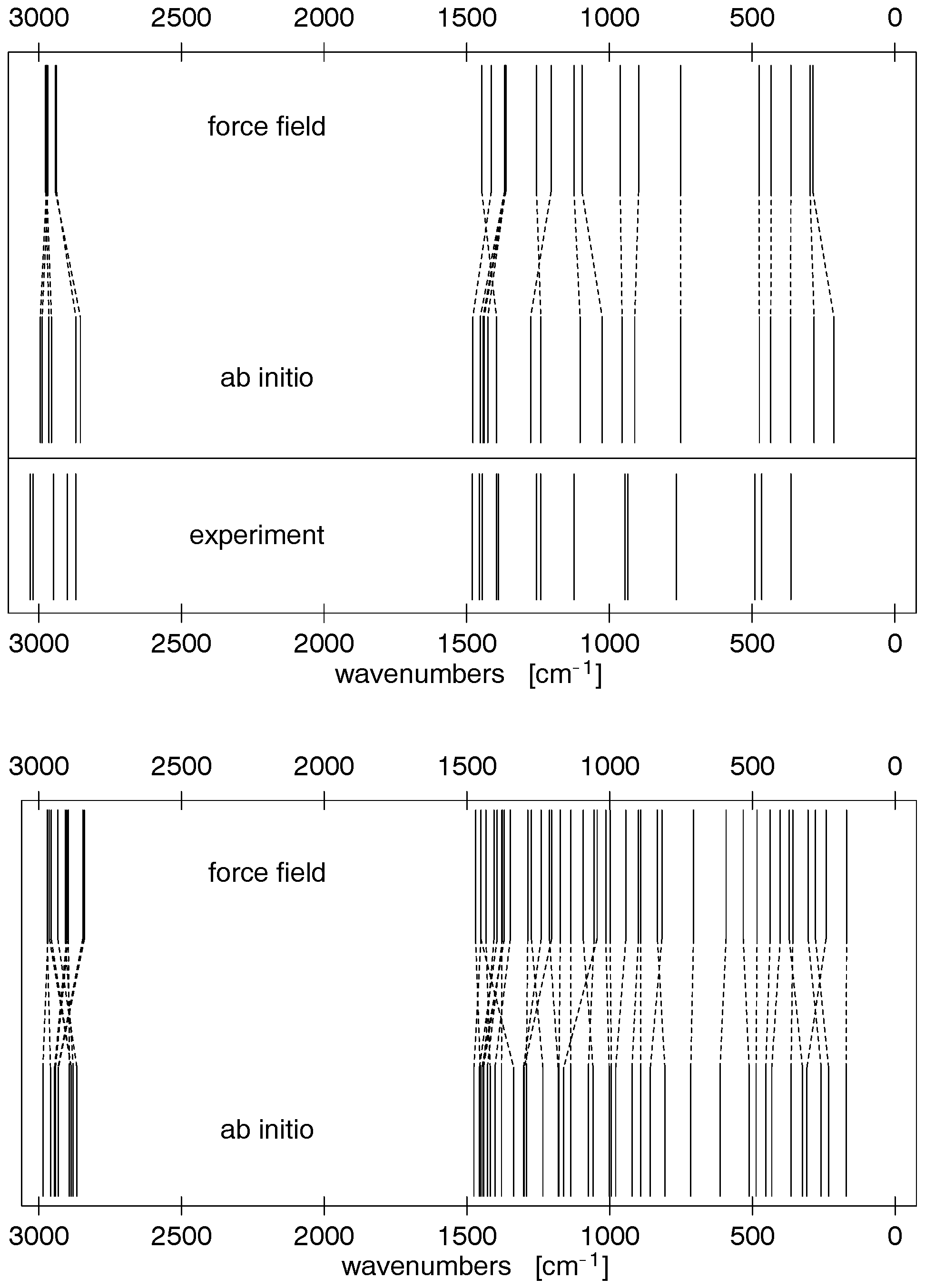 5346 J. Phys. Chem. A, Vol. 107, No. 27, 2003
5346 J. Phys. Chem. A, Vol. 107, No. 27, 2003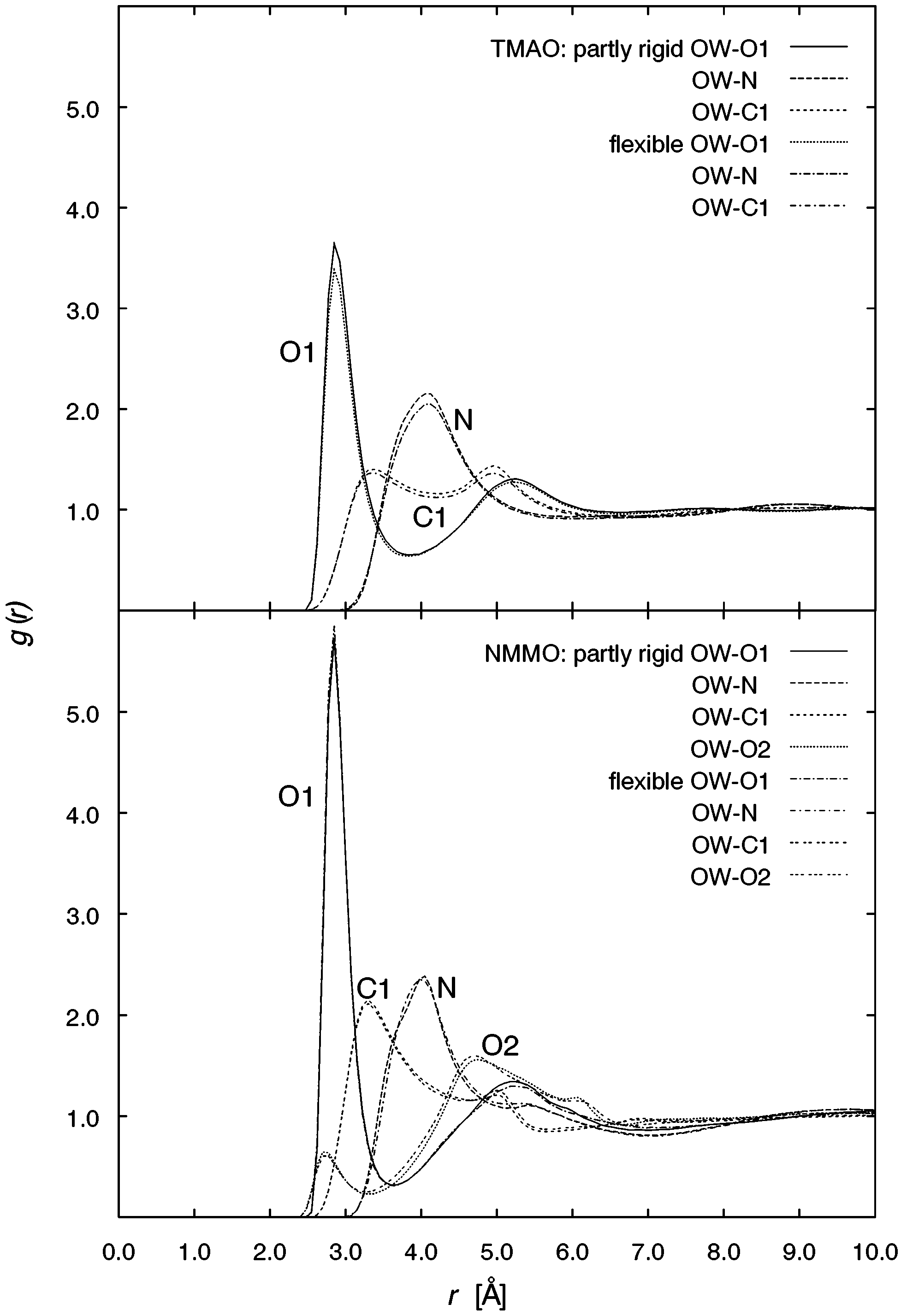
 5348 J. Phys. Chem. A, Vol. 107, No. 27, 2003
5348 J. Phys. Chem. A, Vol. 107, No. 27, 2003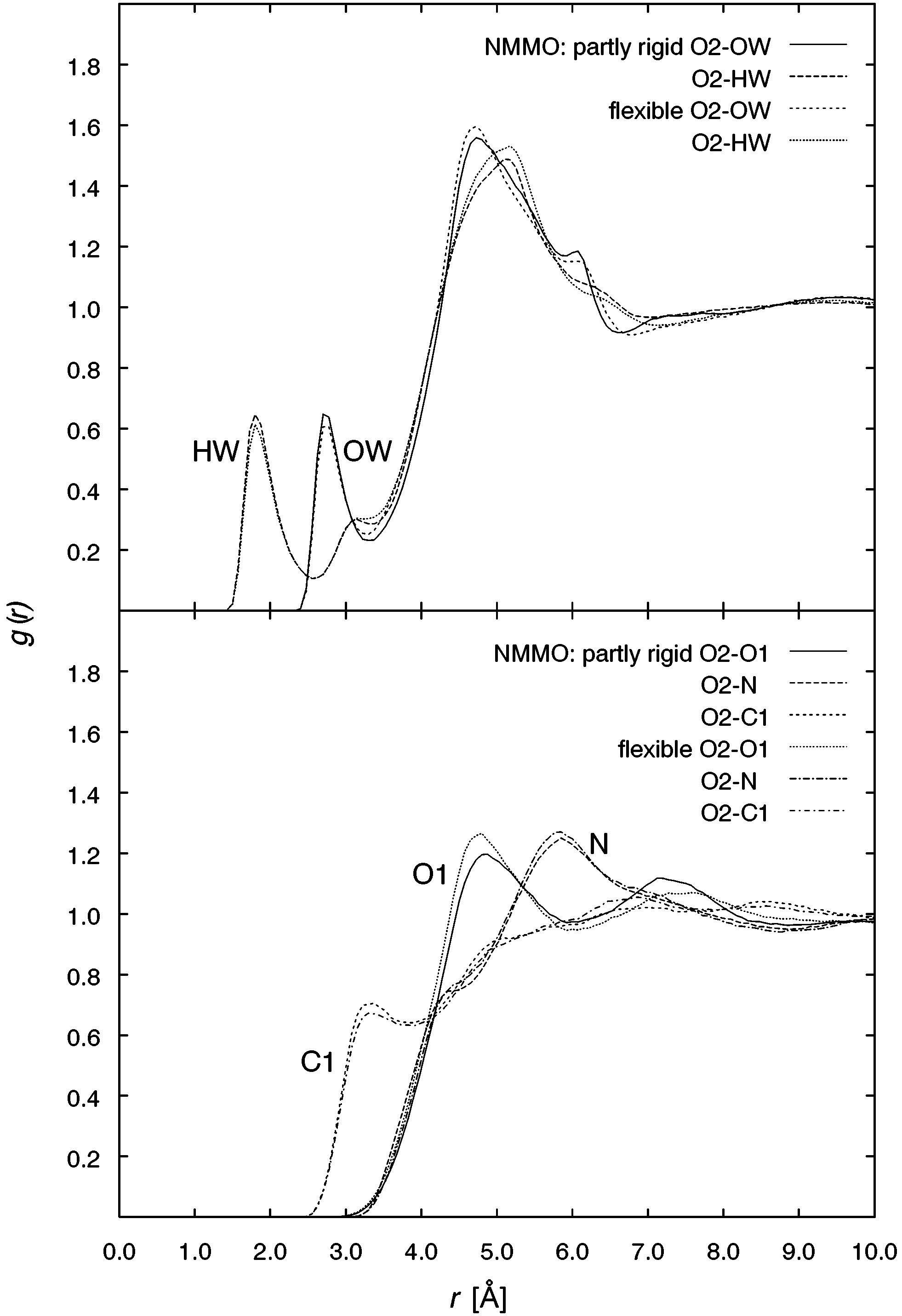
 Binary Phases of Aliphatic N-Oxides and Water
J. Phys. Chem. A, Vol. 107, No. 27, 2003 5349
Binary Phases of Aliphatic N-Oxides and Water
J. Phys. Chem. A, Vol. 107, No. 27, 2003 5349