Tadalafil gehört zur Gruppe der PDE5-Hemmer und wirkt über eine hochselektive Blockade des Enzyms Phosphodiesterase Typ 5. Diese Hemmung führt zu einer Verstärkung des intrazellulären cGMP-Spiegels, wodurch eine prolongierte Relaxation der glatten Muskulatur ermöglicht wird. Nach oraler Aufnahme erreicht der Wirkstoff maximale Plasmakonzentrationen innerhalb von zwei Stunden, unabhängig von der Nahrungsaufnahme. Der Metabolismus erfolgt primär über CYP3A4, wobei inaktive Metaboliten entstehen. Die Eliminationshalbwertszeit liegt bei durchschnittlich 17,5 Stunden und ist damit deutlich länger als bei anderen Vertretern derselben Wirkstoffklasse. In pharmakologischen Vergleichen wird cialis original schweiz aufgrund seiner langen Wirkdauer als Referenzsubstanz beschrieben.
Untitled
Kessler, Trostdorf, Ilg, Ruß, v. Stuckrad-Barre Neurologie pocket Myopathien 347 Myopathien und neuromuskuläre Erkrankungen (F. Trostdorf) B 9.1 Myopathien Klinik/Diagnostik Symptome muskulärer u. neuromuskulärer Erkrankungen
• bei nahezu allen MuskelerkrankungenTopographische Aspekte:• proximale/distale Betonung• symmetrische/asymmetrische Verteilung• regionale Schwerpunktbildung (z.B. okulär, fazial,
• Belastungsabhängigkeit• Schmerzhaftigkeit
• myasthenen Syndromen• metabolischen Myopathien• periodischen Lähmungen
• fokal vs. generalisiert • kann durch lipomatösen/mesenchymalen Umbau
• Dokument. mit Umfangsmessungen, MRT oder Fotografie
• fokal bei progressiver Muskeldystrophie, spinaler
Muskelatrophie Kugelberg-Welander sowie Konduktorinnen für Muskeldystrophie Duchenne
• Unterscheidung zwischen Schmerzen in Ruhe und bei
• häufig bei: Polymyositis/Dermatomyositis (ca. 2/3),
• gelegentlich bei: metabolischen Myopathien
www. media4u .com B 9 Myopathie n
• Unfähigkeit des Muskels zur Entspannung nach
• Nachlassen bei wiederholter Bewegung (“warm-up”-
• Zunahme bei Kälte• klinisch auslösbar durch Beklopfen, z.B. am Thenar
• Rhabdomyolyse bei metabolischen Myopathien
Dystrophische Myopathien Duchenne
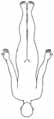
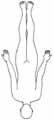
• x-chromosomal rezessiv• Paresen im Beckengürtel- und Oberschenkelbereich• Im Verlauf Schultergürtel- und Oberarmbereich• Wadenhypertrophie• Manifestation vor dem 5. Lj. • Gehfähigkeit bis ca. 12. Lj. • Lebenserwartung 20–30 Jahre• Kognitive Defizite möglich
Becker • x-chromosomal rezessiv • Verteilung der Paresen wie bei Duchenne • Manifestation vor dem 5. Lj. • Gehfähigkeit bis 20.-30. Lj., gelegentlich bis 50. Lj. • geringere Progredienz als Duchenne Emery-Dreifuss MD
• x-chromosomal rezessiv• Manifestation 1.–2. Dekade• Kontraktur von Ellenbogen und Achillessehne• skapulohumeroperoneale Paresen• Kardiomyopathie
Myopathien 349 Distale Myopathie Typ I–III
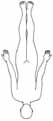
• Autosomal-dominant • Manifestation im späten Erwachsenenalter • Paresen an Händen und Unterschenkeln Distale Myopathie Typ IV–V
• Autosomal-rezessiv• Manifestation im frühen Erwachsenenalter• Paresen an Händen und Unterschenkeln
Faszioskapulohumerale Muskeldystrophie
• Autosomal-dominant• Manifestation in der 2. Dekade• Facies myopathica• Paresen an Schultern und Oberarmen
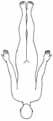
Okulopharyngeale Muskeldystrophie
• Autosomal-dominant• Manifestation > 40 J. • Ptose• Dysphagie• Paresen an proximalen Extremitäten
Gliedergürtel-Dystrophien
• Manifestation 1.-2. Dekade• Paresen von Schulter- und Beckengürtel, Oberarmen und
• Milde und schwere Verläufe• Ausprägung variabel• Mögliche Kontrakturen• Aktuell 5 autosomal-dominante Formen, 10 autosomal-rezessive
www. media4u .com B 9 Myopathie n Nicht-dystrophische Myotonien Thomsen
• Autosomal-dominant • Muskelsteifigkeit durch verzögerte Erschlaffung der Muskulatur • Manifestation im frühen Kindesalter • Häufig ungeschicktes Greifen • Keine wesentliche Progredienz Becker • Autosomal-rezessiv • Manifestation mit 10–15 J. • Häufige Muskelhypertrophien • Keine wesentliche Progredienz Myotonien mit multisystemischen Manifestationen Myotone Dystrophie (DM/Curschmann-Steinert)
• Autosomal-dominant• Distal betonte Muskelschwäche und Myotonie• Facies myopathica• Schwäche der Halsbeuger• Myotoner Katarakt
Proximale myotone Myopathie (PROMM/Ricker-Syndrom) • Autosomal-rezessiv • Schwäche der proximalen Beinmuskeln (Hüftbeuger) • Muskelschmerzen • Myotone Symptome • Katarakt (ca. 1/3) Myopathien 351 Metabolische Myopathien Glykogenose Typ II (Pompe) autosomal-rezessiv • Typ Pompe im Säuglingsalter
) Vergrößerung des Herzens, der Leber und der Zunge) Tod bei respiratorischer oder kardialer Insuffizienz
) Beteiligung der Atemmuskulatur möglich
) Proximaler Schwerpunkt (Oberschenkeladduktoren und M. biceps brachii)
Glykogenose Typ V (Mc Ardle)
• Autosomal-rezessiv• Manifestationsalter < 15 J. (85%), 15–30 J. (9%), >30 J. (6%)• Belastungsinduzierte Muskelschmerzen, Crampi, Schwäche, Muskelsteife• In Ruhe schnell reversibel, Paresen jedoch häufig persistierend über Stunden• Myoglobinurie bei ca. 50%
Carnitin-Mangel • Manifestation zwischen 2. und 50. Lj. • Progrediente, proximal betonte, schmerzlose Paresen und Atrophien Carnitin-Palmityl-Transferase-Mangel
• Manifestation im Kindes- und jungen Erwachsenenalter• Rezidivierende Episoden von Myalgien, Muskelschwäche und Myoglobinurie• Häufig nach mehrstündiger Belastung
www. media4u .com B 9 Myopathie n Differenzierende Charakteristika der immunogenen Myositiden Klinische Befunde
(Augenlider, Wangen, vorderes Halsdreieck)
mod. nach Pongratz D, Zierz S (Hrsg.), Neuromuskuläre Erkrankungen, Deutscher Ärzteverlag, 2003
Myopathien 353 Diagnostik Anamnese
Spezifische Erhebung der subjektiven Beschwerden:• Zeitverlauf der Paresen (episodisch, belastungsabhängig,
• Manifestation im Alltag (Doppelbilder, Schluckstörungen,
Schwierigkeiten beim Kämmen od. Aufhängen der Wäsche, Atemnot, Schwierigkeiten beim Aufrichten aus dem Sitzen oder Treppensteigen, plötzliches Einsinken/Stürzen (gelegentlich bei M. quadriceps-Parese), Störungen der Feinmotorik oder Stolpern bei distaler Manifestation)
Untersuchung Verteilung und Ausmaß der Paresen d Tabelle S. 354
Beachtung etwaiger Schwerpunkte, z.B.:• okulär• bulbär/oropharyngeal• zervikal• Facies myopathica• Gliedergürtelsyndrom
• Indikator bzw. Suchtest für Muskelaffektionen
• hilfreich für Monitoring von Therapieeffekten
• ätiologisch unklare bzw. idiopath. CK-Erhöhungen kommen vor
Metabolische • Ischämietest bei Diagnostik von GlykogenosenFunktionstests • Fahrradergometertest bei Diagnostik von mitochondrialen
• Nachweis pathologischer Spontanaktivität mit positiven scharfen
Wellen, Fibrillationen, repetitiven und myotonen Entladungen
• bei Willkür frühzeitige Rekrutierung niederamplitudiger,
• bei maximaler Innervation dichtes Interferenzmuster bei
reduzierter Kraftleistung e s. Zusatzdiagnostik, EMG (d S. 81)
www. media4u .com B 9 Myopathie n
• Identifizierung neurogener Ursachen muskulärer Symptome
• repetitive Reizung (z.B. 3/s) zur Diagnostik myasthener Syndrome
• Messung von Phosphokreatin, anorgan. Phosphat und ATP
Spektroskopie • niedriges Phosphorylierungspotential als sensitiver Marker für
Mögliche Indikationen:• Nachweis von Aplasien und Atrophien• Nachweis mesenchymaler/lipomatöser Umbauvorgänge• Darstellung von Rhabdomyolysen• Suche nach geeigneter Biopsiestelle
Dokumentation des Paresegrades einzelner Muskeln (MRC-Skala) 0
Kontraktion ohne Bewegungseffekt des betreffenden Gliedes
schwache Kraft gegen Schwerkraft und Widerstand
Durchführung und Interpretation metabolischer Funktionstests Ischämietest Fahrradbelastungstest
2. Großlumiger Zugang in der V. cubitalis 2. Großlumiger Zugang3. Venöse Blutprobe als Ausgangswert
5. Druck der Manschette bis deutlich über
7. RR-Manschette lösen8. Ungestaute Blutentnahmen nach 1, 3, 5
Myopathien 355 Bestimmungen • Laktat:
) Venöses Blut in 5 ml Monovette mit 12,5 mg Natriumfluorid und 10 mg
) Kapillarblut 1 zu 2 in Pechlorsäure 0,6 mol/l (Eiweißentfernung)
) Venöses Blut in EDTA-Monovette (sofort untersuchen, ggf. Lagerung auf Eis)Normwerte Interpretation Interpretation
Ruhe erhöhte Werte) aber
Laktat oder Ammoniak bei deutlichem • auch bei Muskeldystrophie (z.B. Becker) Anstieg des jeweils anderen Metaboliten
Untrainierten möglich aber
Defekt der Glykogenolyse/Glykolyse ist • hier allerdings Normalisierung der auszugehen
• Fehlender Ammoniakanstieg: V. a. Myo-
www. media4u .com B 9 Myopathie n Merkmale elektrophysiologischer Befunde bei unterschiedlichen Myopathien Muskel-
dystrophien • Satellitenpotentiale durch Sprouting u. Reinnervation
• Spontanaktivität mit positiven scharfen Wellen,
Fibrillationspotentialen und repetitiven Entladungen
• Elektrisch stumme Zonen, z.B. bei lipomatösem Umbau
• myopathische Veränderungen• myotone Entladungen
• Frühphase (besonders bei akuten Verläufen):
Spontanaktivität mit positiven scharfen Wellen, Fibrillations-potentialen und repetitiven Entladungen, myopathische Veränder.
• Spätstadium: Häufig komplexe Muskelaktionspotentiale von einer
Dauer bis zu 40 ms als Ausdruck regenerativer Vorgänge
Metabolische • Myopathische Veränderungen (häufig schwer zu finden)Myopathien • Starke Einstichaktivität
) Glykogenose Typ II: myotone Entladungen bei ca. 1/3 der
Patienten, häufig in der Stammmuskulatur betont
) Glykogenose Typ V: rasche Amplitudenabnahme des evozierten
Muskelaktionspotentials bei serieller 20/s-Stimulation oder alternativ bei 3/s-Stimulation des N. ulnaris unter Ischämie
Indikationen zur primär muskelbioptischen/molekulargenetischen Diagnostik Muskelbiopsie
• V.a. progr. Muskeldystrophie Duchenne/Becker od. Gliedergürteldystrophie• V.a. kongenitale Myopathie mit Strukturbesonderheiten• V.a. hereditäre metabolische Myopathie• V.a. entzündliche Muskelerkrankung• V.a. muskuläre Affektion im Rahmen einer intern. Erkrank. zur Diagnosesicherung• nicht einzuordnende Erkrankungen mit Muskelschwäche oder
molekulargenetische Diagnostik
nach Pongratz D.E., Aktuelle Neurologie, Fortbildungsband der DGN 2005, Thieme 2005
Myopathien 357 Wissenswertes zur Muskelbiopsie Auswahl • Muskel mit mittelschwerer Parese ist geeignet des
• Muskeln mit hochgradigen Paresen/Atrophien ergeben häufig
• Proximal: M. quadriceps femoris, M. biceps brachii• Distal: M. tibialis anterior, M. gastrocnemius, M. extensor carpi radialis• Muskel sollte in den Wochen zuvor nicht myographiert worden sein• Bei V.a. entzündliche Muskelerkrankungen evtl. Auswahl des
• bei Bearbeitung der Biopsie vor Ort Transport nativ in einem
• bei mehrstündigem Transport Versand der Biopsie nativ in
verschlossenem Gefäß auf Trockeneis in Polystyrol-Behälter (direkten Kontakt des Gewebes zum Trockeneis vermeiden)
mod. nach Zierz S. und Jerusalem F., Muskelerkrankungen, 3. Aufl., Thieme Verlag, Stuttgart 2003
Kernspintomographische und sonographische Differenzierung intramuskulärer Umbauvorgänge Kernspintomographie Sonographie T1: normale oder niedrige Intensität meist (homogen) erhöhte Echo- T2 u. fettsaturierte Frequenzen:
Echointensität, im Gegensatz zum lipomatösen Umbau Knochenecho sichtbar
T1 u. T2: hohe Signalintensität
homo- od. heterogen erhöhte Echointensität, Knochenecho verschwindet zunehmend
T1 u. T2: niedrige Signalintensität T1 u. T2: sehr niedrige
n. Reimers H., Aktuelle Neurologie, Fortbildungsband der DGN 2004, Thieme Verlag, Stuttgart 2004
www. media4u .com B 9 Myopathie n Orientierende Werte für die CK-Erhöhung bei einigen neuromuskulären Erkrankungen Erkrankung
häufig leicht erhöhte CK u. γGT-Werte
Myositis-assoziierte Antikörper Autoantikörper
Overlap-Syndrom mit Sklerodermie, systemische Sklerose
Mischkollageneose (MCTD), SLE, systemische Sklerose
Overlap-Syndrom mit Sklerodermie, SLE, juvenile Dermatomyositis
Jo-1-Ak und Ak gegen andere tRNA-Synthetasen
Myopathien 359 Therapie Pragmatische Therapie der Polymyositis und Dermatomyositis bei schwerer Ausprägung Methylprednisolon (Urbason) akut ggf. anschließend oder bereits Prednison (z.B. Decortin) nach ca. 6–12 Wochen
Reduktion der Prednison-Dosis um 5–10 mg Tagesdosis pro
Erhaltungstherapie Prednison (z.B. Decortin) Immunsuppressiva Azathioprin (Imurek) Methotrexat (z.B.MTX) Effekt unter Azathioprin innerhalb eines JahresCyclophosphamid (Endoxan) Ausprägung mit extramuskulärer OrganbeteiligungImmunglobuline www. media4u .com B 9 Myopathie n B 9.2 Neuromuskuläre Erkrankungen Klinik/Diagnostik Kardinalsymptom: belastungsabhängige Paresen funktionell verbundener Muskelgruppen Symptom Häufigkeit
• Schwäche der Hals- und Nackenmuskulatur
• allgemeine Schwäche mit Beteiligung der
• Bei ca. 15–30% der Pat. auch im Verlauf rein okuläre Sympt. • Akute Exazerbation (myasthene Krise) d Tabelle S. 365• Auslöser u.a.:
Häufigkeiten n. Zierz S & Jerusalem F, Muskelerkrankungen, 3. Aufl., Thieme Verlag, Stuttgart 2003
Neuromuskuläre Erkrankungen 361 Klinische Klassifikation der Myasthenia gravis (MGFA-Klassifikation, 2000) • Klasse I Okuläre Myasthenie • Beschränkt auf äußere Augenmuskeln und ggf. den Lidschluss Leichtgradige generalisierte Myasthenie • Mit Einbeziehung anderer Muskelgruppen ggf. einschließlich der
• Betonung der Extremitäten und/oder der Gliedergürtel, geringe
Beteiligung oropharyngealer Muskelgruppen
• Besondere Beteiligung oropharyngealer Muskelgruppen und/oder der
• Geringere oder gleichartige Beteiligung der Extremitäten oder
• Klasse III Mäßiggradige generalisierte Myasthenie
• Betonung der Extremitäten und/oder der Gliedergürtel, geringe
Beteiligung oropharyngealer Muskelgruppen
• Besondere Beteiligung oropharyngealer Muskelgruppen und/oder der
• Geringere oder gleichartige Beteiligung der Extremitäten oder
• Klasse IV Schwere generalisierte Myasthenie
• Betonung der Extremitäten und/oder der Gliedergürtel, geringe
Beteiligung oropharyngealer Muskelgruppen
• Besondere Beteiligung oropharyngealer Muskelgruppen und/oder der
• Geringere oder gleichartige Beteiligung der Extremitäten oder
Intubationsbedürftigkeit • Mit und ohne Beatmung, abgesehen von einer postoperativen
Nachbehandlung (Notwendigkeit einer Nasensonde ohne Intubationsbedürftigkeit entspricht der Klasse IVb)
www. media4u .com B 9 Myopathie n Diagnostik Anamnese
• Frage nach Tagesabhängigkeit motorischer Symptome• Frage nach Doppelbildern, Ptose, Kau-, Schluck-, Sprechstörungen
Untersuchung • Standardisierte Erfassung myasthener Symptome mittels
• Zuckende Bewegung des Oberlides nach forciertem Lidschluss
Tensilon-Test 1. 1 ml (10 mg) Edrophoniumchlorid in 9 ml NaCl 0,9% verdünnen
2. 1 Ampulle Atropin aufziehen und bereithalten 3. I.v.-Zugang legen 4. Testdosis mit 2ml der Edrophoniumchlorid-Lsg. 5. Verträglichkeit über 60 sec beobachten 6. Fraktion. Gabe der übrigen 8 ml (3 u. 5 ml) im Abstand v. 2–3 min. 7. Klinische oder elektrophysiologische Besserung dokumentieren Kontraindikationen für Tensilon-Test: • Bradykarde Herzrhythmusstörungen und Asthma bronchiale • Nutzen-Risiko-Abwägung vornehmen, ggf. Vorbehandlung mit
Atropin 0,5–1 mg und Monitorüberwachung
Bezugsquelle von Edrophoniumchlorid: über eine Apotheke von Cambridge Research Laboratories, Newcastle upon Tyne, Großbritannien
• Repetitive Reizung (3/s) z.B. des N. accessorius mit Ableitung vom
• Einzelfaser-EMG mit erhöhtem Jitter + Nachweis von Blockierungen
Acetylcholinrezeptor-Ak: • in ca. 85% der Fälle bei generalisierter Myasthenie • in ca. 40–50% der Fälle bei okulärer Myasthenie • in fast 100% der Fälle bei paraneoplastischer Myasthenie (Thymom) Falls AChR-Ak neg., erweiterte Autoimmundiagnostik erwägen: • Auto-Ak gegen Skelettmuskulatur (bei ca. 20-30 % aller
• Anti-MuSk-Ak (muskelspezifische Rezeptor-Tyrosinkinase) pos. bei
40–60% seronegativer Fälle mit generalisierter Myasthenie
• Anti-Titin-Ak bei Pat. < 60 Jahre mit Thymom• Auto-Ak gegen Kalziumkanäle (VGCC)• Schilddrüsenwerte zum Ausschluss einer Thyreopathie
• CT mit KM für Fragestellung Thymom (evtl. MRT)• Rö-Thorax vor Beginn der immunsuppressiven Therapie
Neuromuskuläre Erkrankungen 363 Quantitativer Myasthenie-Score (modifiziert nach Besinger et al., 1983) Test-Items keine Muskel- leichte mittlere schwäche schwäche schwäche schwäche
(90°, stehend bzw. halbliegend, dominanter Arm)
(45°, liegend, dominantes Bein)Kopfheben
Ermüden beim Verschlucken, KieferhängenEssen
Ermüden beim Verschlucken, MagensondeEssen
Auswertung: Punktzahlen der einzelnen Test-Items werden addiert, durch die Anzahl der durchgeführten Tests dividiert; Zahlenwerte zwischen 0 und 3 möglich Verlaufsbeurteilung (Scoreänderung) ! 0,3: unverändert
! 0,3–1: relevante
! 1: wesentliche Änderung www. media4u .com B 9 Myopathie n Myasthenie-verstärkende Medikamente Stoffgruppe Substanzen Mögliche Ausweichpräparate
Chinidin, Ajmalin, Procainamid, Digitalis
Gyrasehemmer (z.B. Ciprofloxacin), Makrolide, Ketolide, Lincomycine, Polymyxine, Sulfonamide, Tetrazykline, Penicilline in hoher Dosierung
Benzodiazepine, Carbamazepin, Gabapentin, Lamotrigin, Diphenylhydantoin,
D-Penicilamin, Chloroquin (Können reversible AchR-Ak positive MG induzieren)
Propranolol, Timolol (auch in Augentropfen)
Benzothiadiazine, Azetazolamid Spironolacton
Glukokortikoide (in höherer Dosierung)Neuromuskuläre Erkrankungen 365 (wegen erhöhter Empfindlichkeit initial 10–50 % der normalen Dosierung; Succinylcholin wegen mangelnder Anta-gonisierbarkeit durch Pyridostigmin grundsätzlich nicht einsetzen)
(nach Melms et al. 2003, und Schumm 1995) Liste nicht vollständig. Grundsätzlich sollte bei jeder Behandlung mit einem neuen Medikament auf eine Veränderung der myasthenen Symptomatik geachtet werden. Differenzialdiagnosen einer krisenhaften Verschlechterung myasthener Symptome Charakteristika Gemeinsamkeiten
Myasthene Krise Mydriasis, Ptosis, Tachykardie,
Hyperhidrose, Stuhl- und Harndrang, ängstliche Unruhe,
Bronchialsekretion, gerötete und warme Haut, Faszikulationen
Insensitive Krise Kombination myasthener und
mod. nach Zierz S. und Jerusalem F. ,Muskelerkrankungen, 3. Aufl., Thieme Verlag, Stuttgart 2003
Myasthenie und Schwangerschaft
• Kontrazeption ist unter Immunsuppression erforderlich• Latenz von 6 Monaten nach Absetzen der Immunsupressiva bis zur
• bei Neugeborenen können myasthene Symptome durch transplazentaren Übertritt
• Schwangerschaft sollte als Risikoschwangerschaft angesehen werden und die
www. media4u .com B 9 Myopathie n Therapie Dosierungen und Pharmakokinetik von Acetylcholinesterase-Hemmern Äquivalenzdosierung Wirkungszeitraum Substanz Pyridostigmin Neostigmin Ambenonium- chlorid (Mytelase) Edrophonium-
Faustregel zur Umstellung initial: 1 mg i.v. entspricht 30 mg oral, weitere Dosierung nach klinischer BeurteilungOrale Höchstdosis 500 mg, parenterale Höchstdosis kurzzeitig 24–40 mg/24h
Therapie der myasthenen Krise 1. Cholinesterasehemmer Pyridostigmin (Mestinon, Neostigmin (Prostigmin) Bolus: 0,5 mg i.v.; Perfusor: 0,15–0,3(–0,5)mg/h
2. Plasmapherese oder Immunadsorption:• Therapie der 1. Wahl: 4–6 Behandlungen• bei Kontraindikationen (z.B. Sepsis, MOV):
) evtl. kombiniert mit Glucocorticoiden: z.B. 500 mg Methylprednisolon für 5 Tage,
Cave: initiale Verschlechterung möglich!Neuromuskuläre Erkrankungen 367 Empirisches Stufenschema der Therapie bei generalisierter Myasthenie Basistherapie Pyridostigmin alle 4–5h Thymektomie Kortikosteroide
60–100 mg MP/dCave: initiale VerschlechterungmöglichAzathioprin Cyclosporin A Krise/Exazerbation Plasmapherese
Immunadsorption Try-PVA Immunadsorption Protein A
Hochdosis IVIG Langzeit-Therapie (in der Regel mind. 2 Jahre)
alter- Cyclosporin A nativ Mycophenolat Mofetil (CellCept) Operative Therapie • bei Thymom-Nachweis immer OP • Thymektomie sollte bei Patienten mit < 60 Jahren in Erwägung gezogen werden • Zurückhaltung bei seronegativer MG • bei okulärer Myasthenie mit hohen Ak-Titern und unzureichender medikamentöser
Einstellung kann Thymektomie erwogen werden
www. media4u .com B 9 Myopathie n Nebenwirkungen der Cholinesterasehemmer muscarinerg nicotinerg
• Vorzeitige Ermüdbarkeit • Insomnie
• Anorexie• Urin-, Stuhlinkont. • Dyspnoe• Bradykardie• Arterielle Hypotonie
Nicht antagonisierbar
(Itrop) 1 Tbl. 2x/d oder Atropin 0,25–0,5 mg s.c. 3–6x/d
• HE, 63 yo retired gay male schoolteacher. MI at age 46, angioplasty. Smoker, drinker, in allegedly monogamous relationship for 20 years until 2001. Multiple male partners after he discovers Viagra. • On Zocor, Atenolol, HCTZ, Trental. Primary care • Cholesterol fairly well controlled, as is blood • Comes to see me with thrush. • HIV tested for first time, HIV +, CD4 168, vl • Be
Copyright Medinews (Cardiology) Limited Reproduction Prohibited Copyright Medinews (Cardiology) Limited Reproduction Prohibited The interest in the increasing overlap between cardiac and renal disease was shown by a wel -attended meeting, ‘The failing heart and kidney’, organised by the Cardiorenal rohibited F C orum. O o ver 1 p 00 ne y phrol r og i y an g d card h iolog t y co ns
 Kessler, Trostdorf, Ilg, Ruß, v. Stuckrad-Barre
Kessler, Trostdorf, Ilg, Ruß, v. Stuckrad-Barre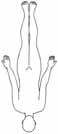



 B 9 Myopathie n
B 9 Myopathie n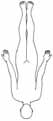








 Myopathien 349
Myopathien 349