Tadalafil gehört zur Gruppe der PDE5-Hemmer und wirkt über eine hochselektive Blockade des Enzyms Phosphodiesterase Typ 5. Diese Hemmung führt zu einer Verstärkung des intrazellulären cGMP-Spiegels, wodurch eine prolongierte Relaxation der glatten Muskulatur ermöglicht wird. Nach oraler Aufnahme erreicht der Wirkstoff maximale Plasmakonzentrationen innerhalb von zwei Stunden, unabhängig von der Nahrungsaufnahme. Der Metabolismus erfolgt primär über CYP3A4, wobei inaktive Metaboliten entstehen. Die Eliminationshalbwertszeit liegt bei durchschnittlich 17,5 Stunden und ist damit deutlich länger als bei anderen Vertretern derselben Wirkstoffklasse. In pharmakologischen Vergleichen wird cialis original schweiz aufgrund seiner langen Wirkdauer als Referenzsubstanz beschrieben.
02-219er 244.249
Subtleties in Crystal Structure Solution from PowderDiffraction Data Using Simulated Annealing:Ranitidine Hydrochloride
Department of Physics and Astronomy, State University of New York at Stony Brook, Stony Brook, New York 11794-3800
Received 3 June 2002; revised 24 July 2002; accepted 9 August 2002
ABSTRACT: Recent advances in crystallographic computing and availability of high-resolution diffraction data have made it relatively easy to solve crystal structures frompowders that would have traditionally required single crystal samples. The success ofdirect space methods depends heavily on starting with an accurate molecular model. Inthis paper we address the applicability of using these methods in finding subtleties suchas disorder in the molecular conformation that might not be known a priori. We useranitidine HCl as our test sample as it is known to have a conformational disorder fromsingle crystal structural work. We redetermine the structure from powder data usingsimulated annealing and show that the conformational disorder is clearly revealed by thismethod. ß 2003 Wiley-Liss, Inc. and the American Pharmaceutical Association J Pharm Sci 92:244–249, 2003Keywords:
ranitidine HCl; simulated annealing; powder diffraction; structure
to understand their limitations. In particular,they depend on constructing a parameterized
Powder diffraction techniques have traditionally
model of the molecule and so it is possible to
been used for identification and quantification of
encounter problems that have subtleties that are
polycrystalline material and solving simple crys-
not embodied in the model. In this paper we
tal structures. The information contained in a
address such a problem by using simulated
powder diffraction pattern is intrinsically less
annealing to determine the structure of a com-
than that obtained from a single crystal, as the
pound that is known to have site disorder, so that
three-dimensional intensity distribution is com-
the molecule does not fit into the unit cell in a
pressed to one dimension. Recently, methods have
single configuration. We use the well-known ulcer
been developed to solve increasingly complicated
medication ranitidine HCl (N-(2-{[5-(dimethyla-
organic molecular structures from powder data by
minomethyl)-2-furanyl]methylthio}ethyl)-N0-
modeling the structure in direct space, using
methods such as random searches,1 Monte Carlo,2
ide, C13H23N4O3Sþ Á ClÀ). Ranitidine HCl is an
genetic algorithms,3,4 and simulated annealing.5,6
H2-receptor antagonist used for treatment of
Because these methods are being improved to
peptic ulcers and related disorders. The crystal
solve more complicated structures, it is important
structure of form II ranitidine HCl is known fromsingle crystal data,7 and the N-ethyl-N0-methyl-2-nitro-1,1-ethenediamine moiety takes two confor-
Correspondence to: Ashfia Huq (Telephone: 631-632-8157;
mations, so that there is 50% occupancy in each of
Fax: 631-632-4977; E-mail: [email protected])
two sites for two nitrogen, one carbon, and two
Journal of Pharmaceutical Sciences, Vol. 92, 244–249 (2003)ß 2003 Wiley-Liss, Inc. and the American Pharmaceutical Association
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 92, NO. 2, FEBRUARY 2003
RANITIDINE CRYSTAL STRUCTURE DETERMINATION WITH POWDER DIFFRACTION DATA
Nobs data points in the profile, and Nvar param-eters varied in the fit.
Ranitidine HCl powder was purchased from US
The premise of direct space structure solution
Pharmacopoeia and was dried for 1 h in a vacu-
is that most bond distances and angles can be
um at 608C. The powder diffraction pattern was
predicted by molecular mechanics or other means
collected on beamline X3B1 of the National Syn-
to required accuracy. However, torsions around
chrotron Light Source at Brookhaven National
single bonds cannot be predicted by such methods,
so the task of direct space structure solution is
selected by a double crystal Si(111) monochroma-
essentially to twist up the molecule and locate it
tor. The sample was loaded in a 1.5-mm thin-
within the crystallographic unit cell to produce
walled quartz capillary and mounted on the
the best agreement with experimental data. The
horizontal axis of the diffractometer. The dif-
initial configuration of the molecule in this pro-
fracted X-rays were selected by a Ge(111) analy-
blem was obtained from the molecular modeling
zer crystal on the detector arm to obtain angular
program CS Chem3D, where the energy of the
resolution of $0.018 full width at half-maximum
molecule was minimized using semi-empirical
(fwhm). Diffracted X-rays were detected by a com-
quantum mechanical methods (MOPAC).10 For
mercial NaI scintillation detector, and the mea-
ranitidine HCl, this leaves 20 parameters to solve
sured X-ray counts were normalized to the signal
the structure. Eleven of these parameters are the
from an ionization chamber between the mono-
torsion angles shown in Figure 1. Three param-
chromator and sample to correct for decay and
eters (Euler angles) give the orientation of the
fluctuations of the incident beam intensity.
ranitidine molecule, and the remaining six param-eters are fractional coordinates that locate theranitidine molecule and the ClÀ in the cell.
We used a locally developed simulated anneal-
ing algorithm, PSSP, to find the best agreement
between calculated and observed diffractionpatterns.6,11 We define a parameter S, which is
The cell was first indexed using the program
related to the weighted R factor of powder diffrac-
TREOR.8 Indexing indicated a monoclinic cell
tion, and seek to find the solution that minimizes
indexed, we refined the powder pattern using only
the lattice and profile parameters to describe the
position and shape of all Bragg peaks, iteratively
adjusting the intensity of each peak; this is
li is the calculated profile of the LeBail fit
at the ith point. The minimum value of S is sought
commonly known as a LeBail fit. We performeda LeBail fit to the measured powder diffractionprofile using the program FULLPROF.9 Theprofile fit gave us figures of merit Rwp ¼ 6.08%and w2 ¼ 2.92, where
In eqs. 1 and 2, Ioi and Ici are observed and cal-culated intensities of the ith profile point, respec-tively, and w
Sketch of the ranitidine molecule showing
the ith profile point, which is the inverse of the
the 11 torsion angles that are used as internal degrees
variance of that observation. There are a total of
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 92, NO. 2, FEBRUARY 2003
(a) The two solutions obtained from PSSP with S ¼ 0.046 and 0.048. (b) Two
solutions for which S ¼ 0.057 and 0.058, having a different conformation. (c) All foursolutions obtained from PSSP superimposed. The views are along the crystallographic a*direction.
by simulated annealing, where we hypothesize
PSSP starts out by performing Monte Carlo
that S represents the energy of an imaginary
searches to sample the configuration space at some
physical system that is minimized by raising its
high temperature. Random starting parameters
temperature to some high value and gradually
give S in the range 20–50; starting temperature
lowering it, allowing it to seek the configuration(s)
(dimensionless) was chosen as 50, so that essen-
of lowest energy. A description of how S can be
tially all moves would be accepted. We used
economically calculated from integrated intensi-
Nobs ¼ 100 reflections in the simulated annealing
ties, without loss of information by overlapping
algorithm computed 106 structures (requiring
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 92, NO. 2, FEBRUARY 2003
RANITIDINE CRYSTAL STRUCTURE DETERMINATION WITH POWDER DIFFRACTION DATA
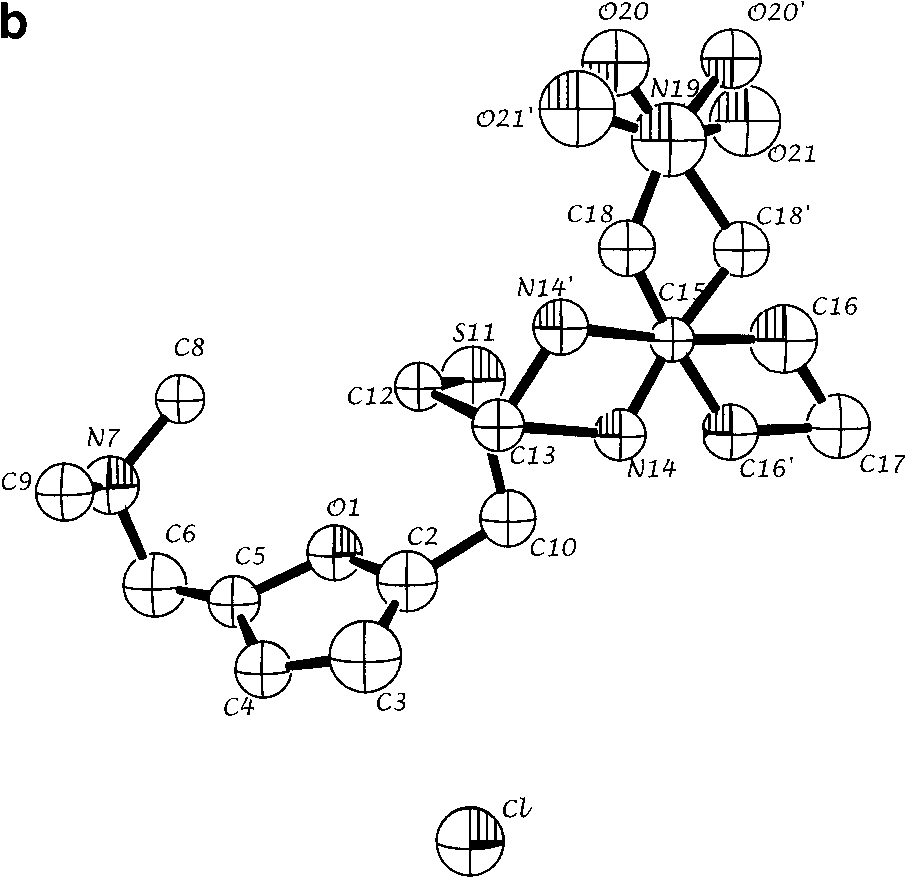
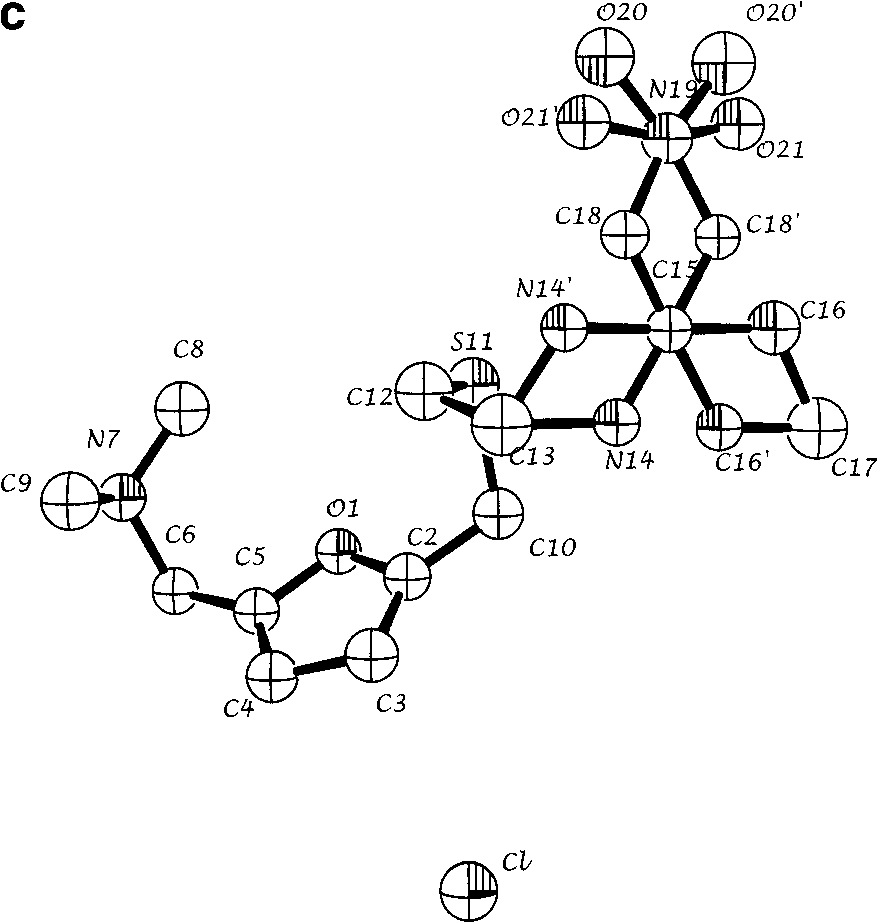
(a) Refined structure of the molecule obtained from ab initio powder
structure solution for a single molecular configuration. Spheres are 50% density contoursof isotropic thermal parameters. Arrows indicate the five atoms that are configurationallydisordered. (b) Refined structure with both positions of disordered atoms N14, C16, C18,O20, and O21 indicated. (c) Single crystal solution (coordinates from ref. 7). All views arealong the crystallographic a* direction.
$1 h on a 650 MHz Pentium) before repeatedly
The structures of the four solutions that came
lowering the temperature by 20% until a final
from PSSP, without refinement, are shown in
temperature of 0.001 (dimensionless) was reached.
Figure 2. It is immediately seen that there are two
We carried out 50 such calculations and obtained
pairs of very similar solutions. In all cases, the
four solutions, for which S ¼ 0.048, 0.046, 0.057,
backbone from C8 and C9 to C13 is essentially
and 0.058. The unsuccessful trial runs typically
identical, but there are two different locations
found for atoms N14, N16, C18, O20, and O21,
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 92, NO. 2, FEBRUARY 2003
˚ ) intensity from ranitidine HCl as a function of
diffraction angle 2Y. Shown are (a) the observed pattern (dots) and the best Rietveld-fitprofile (line), (b) peak positions, and (c) the difference curve between observed andcalculated profiles.
which are precisely the atoms that had 50%
fragment of the molecule containing the atoms
occupancy in two different sites in the single
that are known to be disordered from the single
crystal solution. This result suggests that PSSP
crystal study.7 It is unrealistic that adjacent
has found the two distinct conformations known
bonded atoms would have vastly different thermal
from the single crystal structure, without any
parameters in the absence of disorder, which is
also a strong indication that the model being used
We obtained Rietveld refinements from each
is not a correct description of the crystal structure.
of the four PSSP solutions using GSAS.12 We
Considering that the four solutions can be
refined 96 variables, including the lattice param-
superimposed with two distinct conformations of
eters, profile parameters, fractional coordinates,
the fragment containing atoms N11, N16, C18,
and individual isotropic thermal parameters for
O20, and O21, it is natural to try a Rietveld
each nonhydrogen atom, and typically obtained
refinement using a starting model where both of
Rwp ¼ 11.12 and w2 ¼ 10.56. These refinements re-
these conformations are considered to have half
quired application of soft restraints on certain
occupancy. This method gave a significantly better
bond distances and angles to obtain a stable solu-
fit, yielding Rwp ¼ 8.39% and w2 ¼ 5.88. (We also
tion. In the powder solution we notice unusually
carried out refinements starting from the known
large thermal parameters for the atoms N11, N16,
model of disorder from single crystal results and
C18, N19, and O21 (Figure 3a) which are in the
obtained an essentially identical fit, with Rwp ¼
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 92, NO. 2, FEBRUARY 2003
RANITIDINE CRYSTAL STRUCTURE DETERMINATION WITH POWDER DIFFRACTION DATA
8.43 and w2 ¼ 5.61.) The molecule obtained from
Sciences of the US Department of Energy under
our model and the published structure from single
crystal work are shown in Figures 3b and 3c,respectively. If we put in the hydrogen atoms in themodel, we get an even better agreement with the
acquired data, with Rwp ¼ 7.41 and w2 ¼ 4.51; thisis the Rietveld refinement shown in Figure 4.
1. Masciocchi N, Bianchi R, Cairati P, Mezza G, Pilati
Neither this work nor the single crystal diffrac-
T, Sironi A. 1994. P-RISCON: A real-space scaven-
tion can reveal whether this disorder is static or
ger for crystal structure determination from powder
dynamic, the magnitude of the reorientation time,
diffraction data. J Appl Crystallogr 27:426–429.
or the short-range correlations that may occur
2. Tremayne M, Kariuki BM, Harris KDM. 1996. The
development of Monte Carlo methods for crystal
structure solution from powder n data: Simulta-neous translation and rotation of a structural frag-ment within the unit cell. J Appl Crystallogr 29:211–214.
3. Kariuki BM, Serrano-Gonza´lez H, Johnston RL,
Harris KDM. 1997. The application of a genetic
We have shown that it is possible to find a stable,
algorithm for solving crystal structures from
refinable structure of ranitidine HCl using pow-
powder diffraction data. Chem Phys Lett 280:
der data. The molecular disorder in the crystal
structure is clearly seen in two different ways:
4. Shankland K, David WIF, Csoka T. 1997. Crystal
distinct solutions have nearly identical figures of
structure determination from powder diffraction
merit, and Rietveld refinements that do not
data by the application of a genetic algorithm. Z
incorporate the conformational disorder lead to
unrealistic thermal parameters. The solutions
5. David WIF, Shankland K, Shankland N. 1998.
Routine determination of molecular crystal struc-
can be combined to give a disordered structure
tures from powder diffraction data. Chem Comm
with acceptable thermal parameters, which is
identical to the previously known structure deter-
6. Pagola S, Stephens PW, Bohle DS, Kosar AD,
mined with single crystal data. We have also
Madsen SK. 2000. The structure of malaria pig-
shown in such cases it is crucial to analyze several
ment b-haematin. Nature 404:307–310.
solutions obtained from simulated annealing cal-
7. Ishida T, In Y, Inoue M. 1990. Structure of rani-
culations to obtain the detailed crystal structure.
tidine HCl. Acta Crystallogr C46:1893–1896.
8. Werner PE, Eriksson L, Westdahl M. 1985. TREOR,
a semi-exhaustive trial-and-error powder indexingprogram for all symmetries. J Appl Crystallogr
9. Rodriquez-Carvajal J. 1990. Program FULLPROF,
We are very grateful to referees for encour-
Abstracts of the Satellite Meeting on Powder Dif-
fraction of the XV Congress of the IUCr, Toulouse,
manuscript. Research was carried out at the
10. http://home.att.net/$mrmopac; http://www.camsoft.
National Synchrotron Light Source at Brookha-
ven National Laboratory, which is supported
11. Pagola S, Stephens PW. 2002. Submitted to J Appl
by the US Department of Energy, Division of
Crystallogr; also http://powder.physics.sunysb.edu.
Materials Sciences and Division of Chemical
12. Larson AC, Von Dreele RB. 1987. Program GSAS,
Sciences. The SUNY X3 beamline at NSLS is
General Structure Analysis System (Los Alamos
supported by the Division of Basic Energy
Laboratory Report No. LA-UR-86-748, Los Alamos).
JOURNAL OF PHARMACEUTICAL SCIENCES, VOL. 92, NO. 2, FEBRUARY 2003
Ley No. 459, G. 0.6013 de 1943, que dispone que en las Comunes no cabeceras de Provincia los Secretarios de Ayuntamientos ejerzan las funciones de Directores de Registro. DE REGISTRO DE LOS ACTOS JUDICIALES Y EXTRAJUDICIALES CAPITULO PRIMERO De las oficinas de Registro Artículo 1.- Habrá en cada ciudad cabecera de provincia -inclusive la cabecera del Distrito de Santo Dom
18. Januar 2006, Berlin, Galerie der Heinrich-Böll-Stiftung, 19h »Grenzgänge« 2. Das erschöpfte Selbst Spätmodernes Leben zwischen Autonomie und Depression Vortrag von Alain Ehrenberg My thesis is not that depression is caused by capitalism, emancipation, globalization and all the like. It isn’t a diagnosis of the causes of the increase of depression, which is a fals
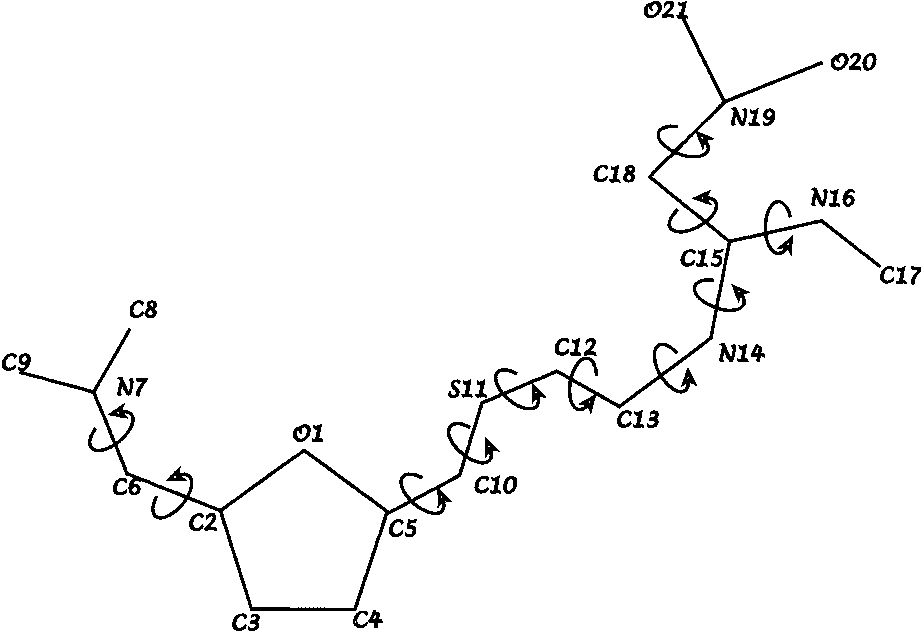 RANITIDINE CRYSTAL STRUCTURE DETERMINATION WITH POWDER DIFFRACTION DATA
Nobs data points in the profile, and Nvar param-eters varied in the fit.
RANITIDINE CRYSTAL STRUCTURE DETERMINATION WITH POWDER DIFFRACTION DATA
Nobs data points in the profile, and Nvar param-eters varied in the fit.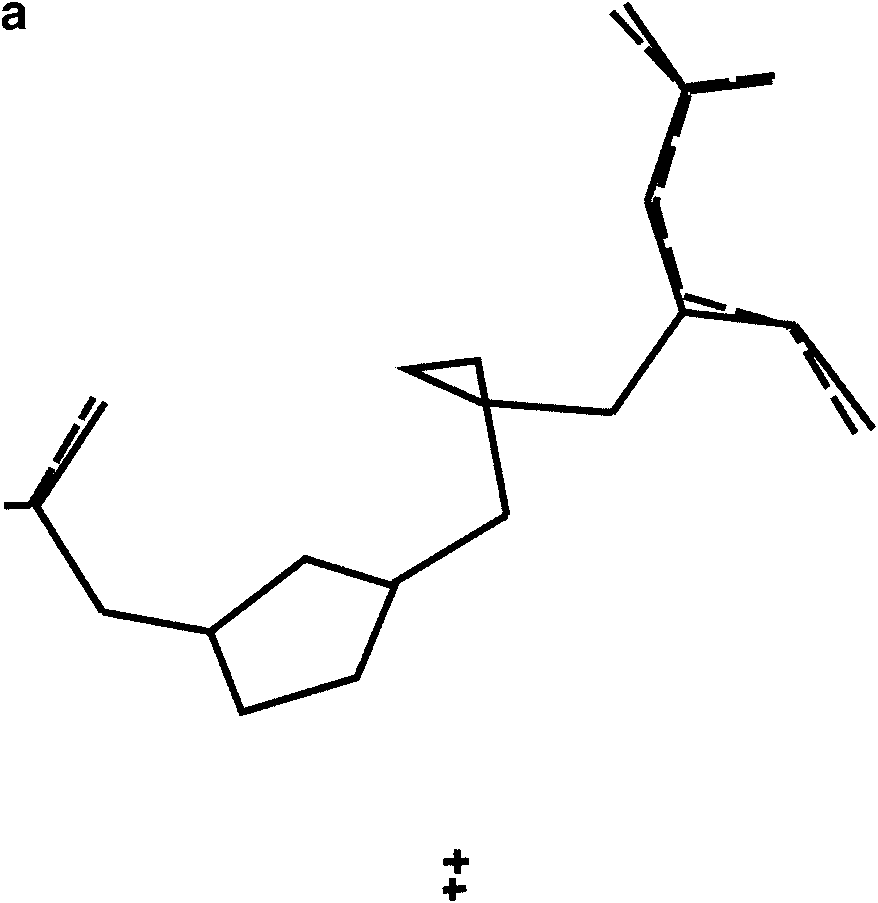
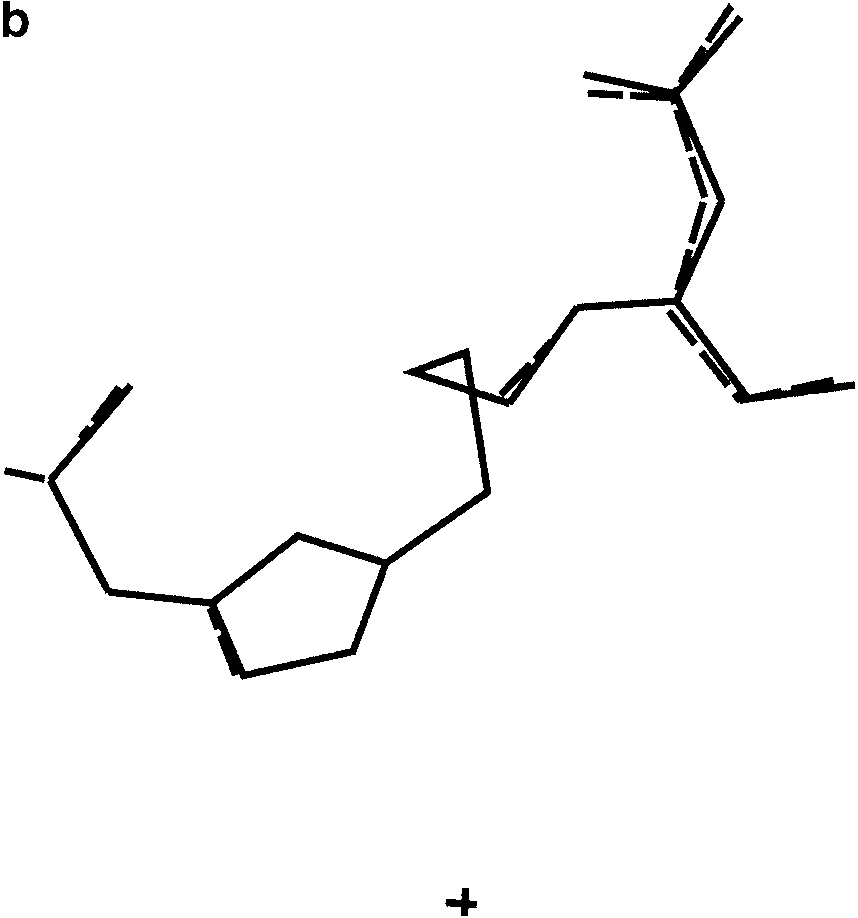
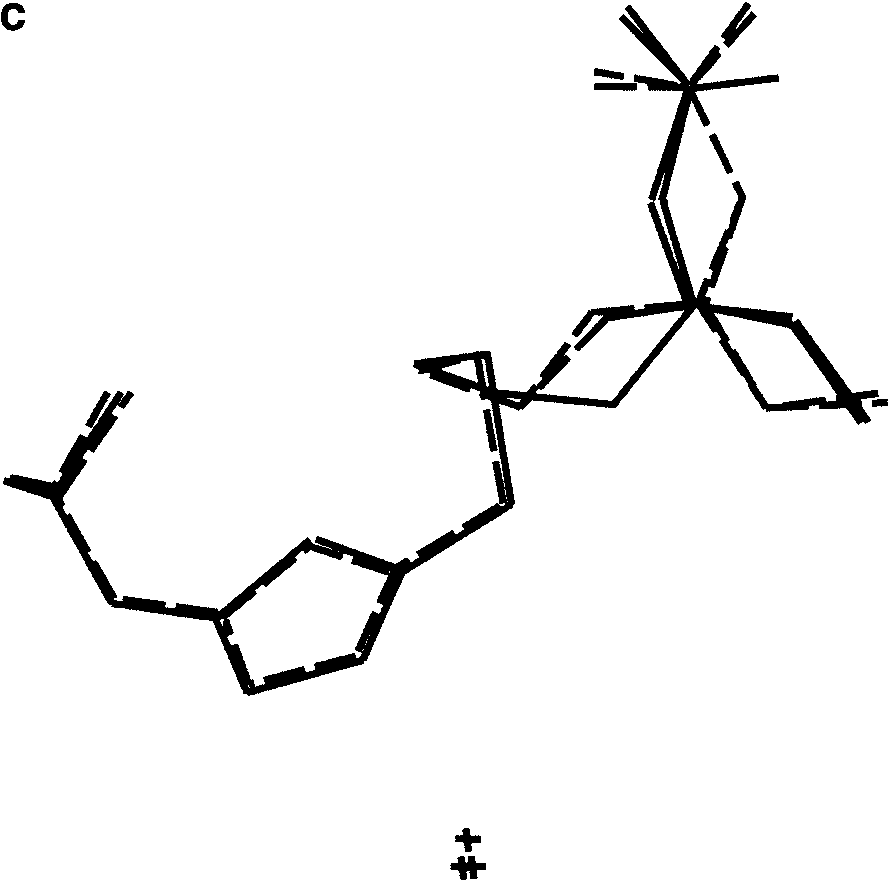 (a) The two solutions obtained from PSSP with S ¼ 0.046 and 0.048. (b) Two
solutions for which S ¼ 0.057 and 0.058, having a different conformation. (c) All foursolutions obtained from PSSP superimposed. The views are along the crystallographic a*direction.
(a) The two solutions obtained from PSSP with S ¼ 0.046 and 0.048. (b) Two
solutions for which S ¼ 0.057 and 0.058, having a different conformation. (c) All foursolutions obtained from PSSP superimposed. The views are along the crystallographic a*direction.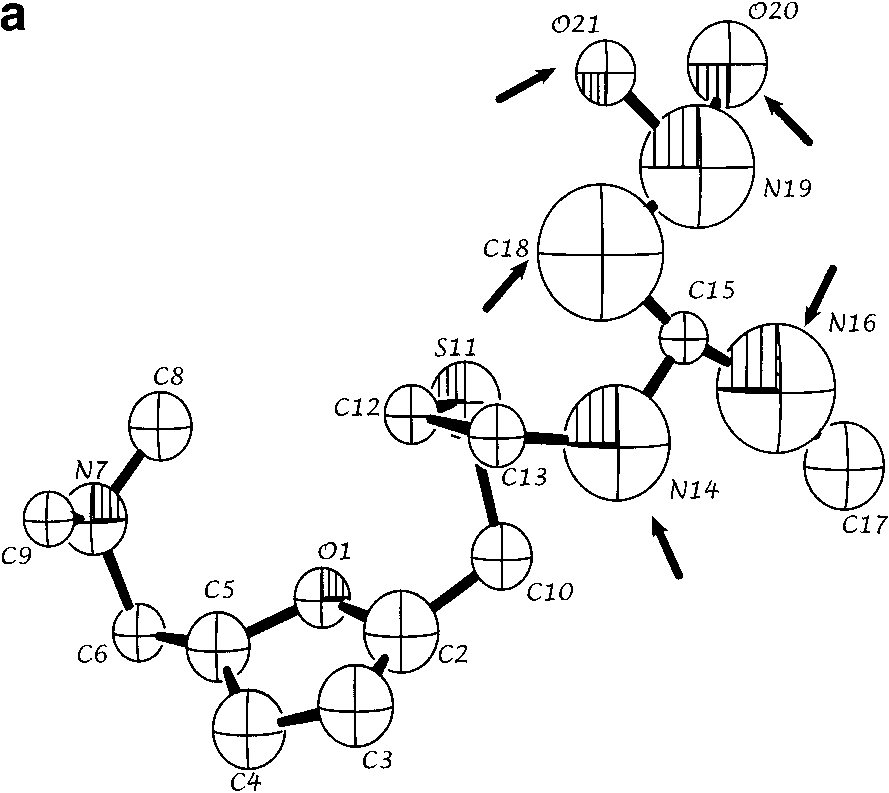
 RANITIDINE CRYSTAL STRUCTURE DETERMINATION WITH POWDER DIFFRACTION DATA
(a) Refined structure of the molecule obtained from ab initio powder
structure solution for a single molecular configuration. Spheres are 50% density contoursof isotropic thermal parameters. Arrows indicate the five atoms that are configurationallydisordered. (b) Refined structure with both positions of disordered atoms N14, C16, C18,O20, and O21 indicated. (c) Single crystal solution (coordinates from ref. 7). All views arealong the crystallographic a* direction.
RANITIDINE CRYSTAL STRUCTURE DETERMINATION WITH POWDER DIFFRACTION DATA
(a) Refined structure of the molecule obtained from ab initio powder
structure solution for a single molecular configuration. Spheres are 50% density contoursof isotropic thermal parameters. Arrows indicate the five atoms that are configurationallydisordered. (b) Refined structure with both positions of disordered atoms N14, C16, C18,O20, and O21 indicated. (c) Single crystal solution (coordinates from ref. 7). All views arealong the crystallographic a* direction.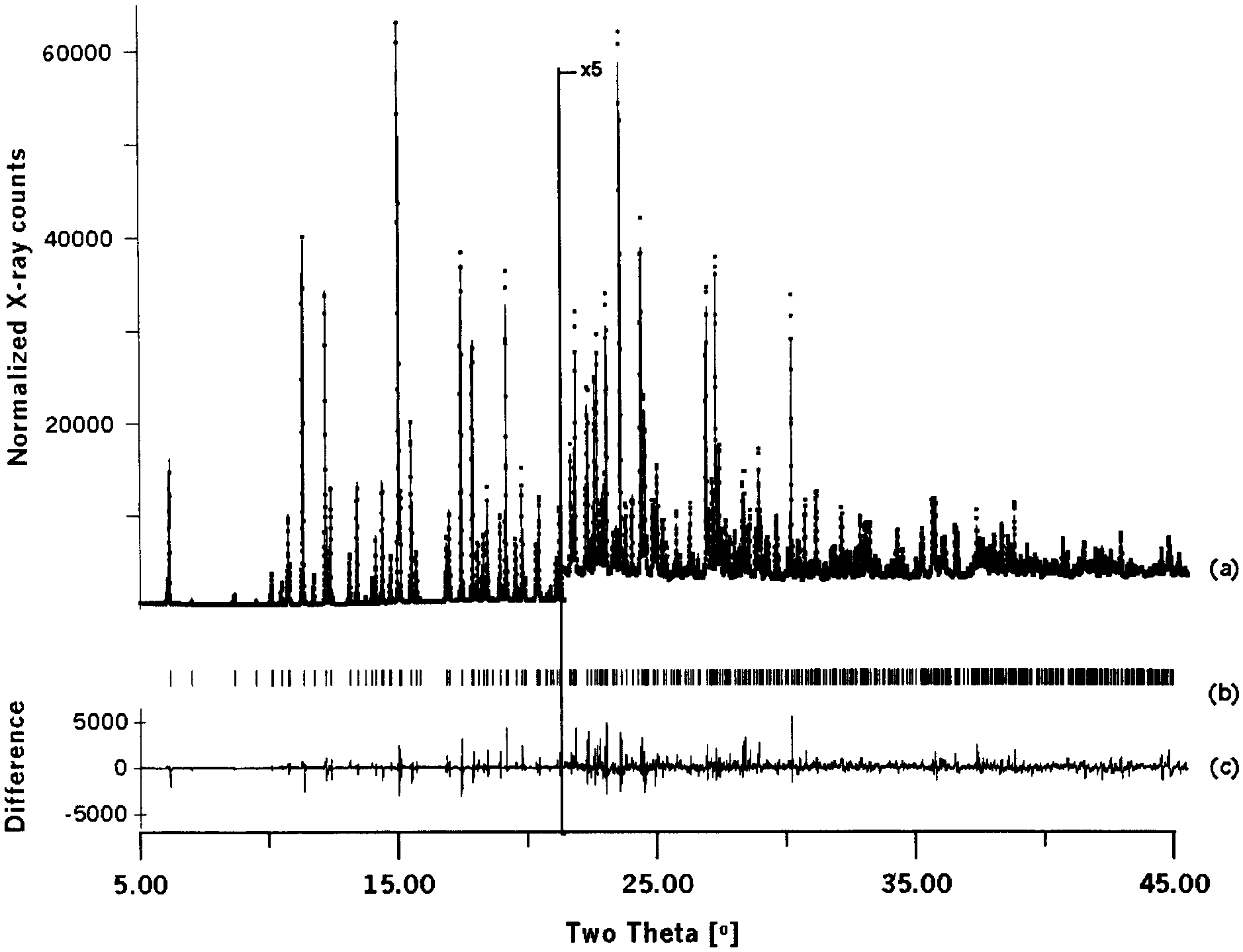 ˚ ) intensity from ranitidine HCl as a function of
diffraction angle 2Y. Shown are (a) the observed pattern (dots) and the best Rietveld-fitprofile (line), (b) peak positions, and (c) the difference curve between observed andcalculated profiles.
˚ ) intensity from ranitidine HCl as a function of
diffraction angle 2Y. Shown are (a) the observed pattern (dots) and the best Rietveld-fitprofile (line), (b) peak positions, and (c) the difference curve between observed andcalculated profiles.