Tadalafil gehört zur Gruppe der PDE5-Hemmer und wirkt über eine hochselektive Blockade des Enzyms Phosphodiesterase Typ 5. Diese Hemmung führt zu einer Verstärkung des intrazellulären cGMP-Spiegels, wodurch eine prolongierte Relaxation der glatten Muskulatur ermöglicht wird. Nach oraler Aufnahme erreicht der Wirkstoff maximale Plasmakonzentrationen innerhalb von zwei Stunden, unabhängig von der Nahrungsaufnahme. Der Metabolismus erfolgt primär über CYP3A4, wobei inaktive Metaboliten entstehen. Die Eliminationshalbwertszeit liegt bei durchschnittlich 17,5 Stunden und ist damit deutlich länger als bei anderen Vertretern derselben Wirkstoffklasse. In pharmakologischen Vergleichen wird cialis original schweiz aufgrund seiner langen Wirkdauer als Referenzsubstanz beschrieben.
08-arteche.qxp
J. Mex. Chem. Soc. 2005, 49(4), 353-358 Studies on the Selective S-oxidation of Albendazole, Fenbendazole, Triclabendazole, and Other Benzimidazole Sulfides
Olivia Soria-Arteche,1* Rafael Castillo,2 Alicia Hernández-Campos,2 Marcela Hurtado-de la Peña,1Gabriel Navarrete-Vázquez,2, 3 José Luis Medina-Franco,2 Kathia Gómez-Flores.1
1 Departamento Sistemas Biológicos, DCBS. Universidad Autónoma Metropolitana-Xochimilco, México, D.F. 04960, México.
2 Departamento de Farmacia, Facultad de Química, Universidad Nacional Autónoma de México, México D.F. 04510, México3 Facultad de Farmacia, Universidad Autónoma del Estado de Morelos, Cuernavaca, Morelos 62210, México
Recibido el 16 de agosto de 2005; aceptado el 14 de diciembre de 2005
Abstract. The selective S-oxidation of albendazole, fenbendazole, Resumen. La oxidación selectiva de albendazol, fenbendazol, y otros
and other benzimidazole sulfides with sodium periodate in acid medi-
sulfuros bencimidazólicos con peryodato de sodio en medio ácido da
um, afforded the corresponding sulfoxides or sulfones. In contrast,
los correspondientes sulfóxidos y sulfonas. En contraste, triclabenda-
triclabendazole and other 2-methylthiobenzimidazole derivatives
zol y otros derivados de 2-metiltiobencimidazoles no pueden ser oxi-
could not be S-oxidized under the same smooth conditions with this
dados bajo las mismas condiciones suaves con este reactivo, pero sí
reagent, but with MCPBA, a stronger oxidizing agent.
con un agente oxidante fuerte como MCPBA. Keywords: Albendazole, fenbendazole, triclabendazole, metabolites, Palabras clave: Albendazol, fenbendazol, triclabendazol, metaboli- Introduction
sulfones that are difficult to separate. The need of thesemetabolites in helminthiasis chemotherapy research [2,3,5]
A large group of wide spectrum, high efficiency anthelmintics,
makes the development of new preparation methods highly
such as the benzimidazole 2-carbamates (BZC), is marketed
desirable, in particular, those that employ common reagents,
worldwide for the control of helminthiasis. It has been reported
mild reaction conditions and convenient working procedures.
that benzimidazole anthelmintics with a sulfide group are the
In this paper we present an efficient, high yield method for
most active against intestinal nematodes in humans, as well as
the selective S-oxidation of 1, 2 and 3 to obtain 4, 6 and 8, as well
in animals [1-3]. Included among these anthelmintics are alben-
as the selective S-oxidation of other benzimidazole sulfides 10, 12
dazole 1, fenbendazole 2 and triclabendazole 3 (Figure 1).
and 17 to obtain 11, 13 and 18, respectively (cf. Figures 2 and
Benzimidazole sulfides 1, 2, and 3 undergo first pass bio-
Scheme). In these studies, sodium periodate in acid medium was
transformation in the organism, where the sulphur atom is oxi-
used as the oxidizing agent. This reagent does not over-oxidize 1
dized to produce the active antiparasitic sulfoxides 4 [1,4-5], 6
under low temperature conditions [15-17]. In addition, aqueous
[6-7], and 8 [8-9], respectively. Further oxidation produces the
mixtures of acetic acid-acetonitrile were used as solvent, which
inactive sulfones 5, 7 or 9.
allowed carrying out the reactions at different temperature condi-
Metabolites 4, 5 and 7 are commercially available but not
tions for better control, thus avoiding over oxidation.
easily affordable. Not so for 6, which is easily available at a relatively low price. Although there are reports in the pertinent literature for the synthesis of 4, 5[1,10-12]; 6, 7 [2]; and 8, 9 Results and Discussion
[13], in addition to the general methods of S-oxidation [13],these are not easy to carry out, or fail, due to insolubility prob-
The results of the oxidation reactions of 1-3, 10, 12 and 17 are
lems in 1-3, which often leads to mixtures of sulfoxides and Fig. 1. Structure of albendazole 1, fenbendazole 2, triclabendazole 3, and their metabolites. J. Mex. Chem. Soc. 2005, 49(4) Table 1. Oxidation reactions, conditions and results
Oxidation of 1 with sodium periodate in acetic acid to
temperature and the equivalents were increased (60°C, 2.8
obtain albendazole sulfoxide 4 was studied under several tem-
eq.), sulfone 7 was obtained as the only product in a 67%
perature conditions. At -10°C it was necessary to add acetoni-
trile as co-solvent to avoid precipitation of 1 and to complete
In order to increase the solubility of 3 and prevent its pre-
its oxidation; however, the reaction was incomplete. On the
cipitation, the oxidation reaction with sodium periodate was
other hand, at 25°C, a mixture of 1, 4 and 5 was produced.
undertaken with acetonitrile as co-solvent; however, although
The best results were obtained when the reaction was carried
a solution was attained, no change in 3 was observed, even at
out in acetic acid-water at 0-5°C, in this case, 4 was obtained
20°C. In this case, we had to use m-chloroperbenzoic acid
as the only product in a 97% yield. Its 1H NMR spectrum
(MCPBA), a stronger oxidizing agent, and obtained 8 at 0-5°C
showed a multiplet at 2.72-2.86 ppm, characteristic of the
diastereotopic α-methylene hydrogens next to the chiral sul-
In the case of compound 10, the oxidation with sodium
foxide. The mass spectrum showed a peak at m/z 281, which
periodate in acetic acid-acetonitrile proceeded smoothly at 0-
is in agreement with the molecular ion of 4. The purity of 4
5°C to afford sulfoxide 11 in a 90% yield.
was confirmed by HPLC. Only one peak with a 6.75 min
The oxidation of 12 to the sulfoxide 13 also failed with
sodium periodate, but it was easily achieved with MCPBA.
When 1 was oxidized with excess of sodium periodate at
The lower reactivity of sulfides such as 3 and 12 can be attrib-
25°C for longer periods of time, sulfone 5 was the only prod-
uted to a reduced electron density on sulfur because of the
uct obtained in a 90% yield. The 1H NMR spectrum now
inductive effect of the imidazole ring nitrogen atoms. This
showed a triplet at 3.21 ppm for the nondiastereotopic α-meth-
contention is supported by the regiospecific and high yielding
ylene hydrogens next to the sulfone group. The mass spectrum
oxidation of the bis-sulfide 17 (Scheme 1[19, 20]; see
showed the molecular ion peak of 5 at m/z 297. The purity
Experimental section for details of synthesis) to the monosul-
was confirmed by HPLC, a single peak with a 5.21 min reten-
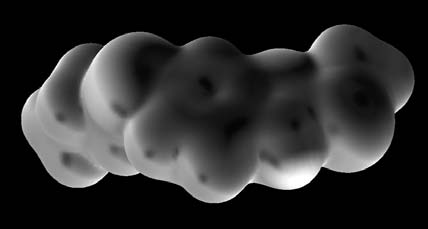
foxide 18, and by electron density calculations (Fig. 3).
Encouraged by these results, we decided to test the perio-
date oxidation method with compounds 2, 3 and other benz- Conclusions
imidazole sulfides, 10 and 12, which are currently being stud- ied as experimental new antiparasitic agents (Figure 2).
A practical, mild and efficient method for the S-oxidation of
Oxidation of 2 at 15°C gave sulfoxide 6 in a 95% yield.
albendazole 1, fenbendazole 2, and benzimidazole sulfide 10
Its structure was confirmed by mass spectrometry. When the
was developed. The method consists in treating a cold solu-
Fig. 2. Benzimidazole sulfides used as experimental antiparasitic agents and their sulfoxides.
Studies on the Selective S-oxidation of Albendazole, Fenbendazole, Triclabendazole, and Other Benzimidazole Sulfides
Scheme 1. Reagents: (a) SnCl2·2H2O, EtOH; (b) CS2, KOH, EtOH; (c) CH3I, KOH, CH3COCH3; (d) NaIO4. Fig. 3. Molecular surface of 17 showing the potential energy calculated at RHF/6-31G(d,p) level. Darker zones represent either more positive or more negative regions. The sulphur atom of the propylthio group (S5, charge -0.366) corresponds to a darker zone than the sulphur atom of the methylthio group (S2, charge -0.211).
tion of these compounds with sodium periodate to generate the
sext, sextuplet; m, multiplet; bs, broad signal. HPLC analyses
corresponding sulfoxides. The related sulfones were obtained
were performed in a Perkin Elmer serie 200LC, UV 785A
at higher temperatures. In the case of 2-(methylthio)benzimi-
detector: column C-8, mobile phase: CH3OH-H2O-CH3CN-
dazoles, such as triclabendazole 3, the S-oxidation was
CH3COOH (40:40:19.4:0.6). Starting materials 1, 2, and 3
achieved with MCPBA, a stronger oxidizing agent.
were obtained commercially, where as 10, 12, and 14 were synthesized in our laboratories. Experimental General method for the synthesis of propylsulfinyl deriva- tives (4, 6, 11 and 18) and propylsulfonyl derivatives (5, 7).
Melting points were determined on a Büchi B-540 melting
Into a stirred solution of 1, 2, 10, or 17 in AcOH or
point apparatus and are uncorrected. Reactions were moni-
AcOH/CH3CN (1:1) was slowly added, dropwise, a solution
tored by TLC on 0.2 mm precoated silica gel 60 F
4 in a mixture of H2O/AcOH. The mixture was stirred,
Merck). Infra-red spectra were recorded in a Perkin-Elmer FT-
then, the solvent removed in vacuo without heating. The
IR-1600 spectrometer on KBr pellets, the absorption bands are
progress of the reaction was monitored by TLC. The residue
given cm-1. MS were recorded on a JEOL JMS-SX102A spec-
was suspended in brine and neutralized with a saturated solu-
trometer by electron impact (EI) of low and high resolution
tion of potassium carbonate, the resulting suspension was fil-
(HR-MS), and FAB+. 1H NMR spectra were measured with a
tered, and the residue washed with water and air dried.
Varian model EM-390 (300 MHz) spectrometer. Chemicalshifts are given in ppm relative to Me
Methyl 5-[(propylsulfinyl)-1H-benzimidazol-2-yl]carbamate
nal standard. The solvent employed was DMSO-d(4). Following the general procedure, 1 (0.5 g, 1.89 mmol,) in 11 and 17 that was CDCl
3. J values are given in Hz. The fol-
lowing abbreviations are used: s, singlet; d, doublet; t, triplet;
H2O/AcOH (5:2) were stirred at 0-5ºC for 2 h and gave 4 J. Mex. Chem. Soc. 2005, 49(4)
(0.514 g, 97%) as a white powder. Mp 218-220ºC. TLC
MS (EI) calcd for C11H11F3N2OS (M+) m/z 276.0544, found:
(Toluene-THF-AcOH, 5:1:1). IR νmax 3169 (NH), 1730
276.0546. 1H-NMR: δ 1.06 (3H, t, J = 7.34, CH3CH2CH2SO),
(C=O), and 1028 (SO). MS EI (m/z): 281 (M+), HRMS (EI)
1.60-1.90 (2H, m, CH2CH2SO), 2.83-3.0 (2H, m, CH2SO),
Calcd for C12H15N3O3S (M+) m/z: 281.0834, found: 281.0820.
7.48 (1H, d, J = 7.9, H-6), 7.89 (1H, d, J = 7.9, H-7), 8.25
1H-NMR: δ 0.95 (3H, t, J= 7.20, CH3CH2CH2SO), 1.42-1.66
(1H, s, H-4), and 10.58 (1H, bs, NH int. D2O).
(2H, m, CH2CH2SO), 2.72-2.86 (2H, m, CH2SO), 3.79 (3H, s,CH3O), 7.33 (1H, dd, J = 8.0, J = 1.4, H-6), 7.57 (1H, d, J =
2-(Methylthio)-5-(propylsulfinyl)-1H-benzimidazole (18).
8.0, H-7), 7.72 (1H, d, J = 1.4, H-4), and 11.90 (H, bs, NH,
Following the general procedure, 17 (0.105 g, 0.44 mmol)) in
10 mL of AcOH/CH3CN and NaIO4 (0.094 g, 0.44 mmol) in 2 mL of H2O/AcOH were stirred for 2 h and gave 18 (0.900 g, Methyl [5-(propylsulfonyl)-1H-benzimidazol-2-yl]carba-
80%) as a white powder, after recrystallization from AcOEt-
mate (5). Following the general procedure, 1 (1.0 g, 3.76
Et2O. Mp: 100.1-100.5 °C. TLC (CHCl3-CH3OH, 95.5:0.5).
mmol) in 15 mL of AcOH and NaIO4 (2.015 g, 9.42 mmol, 2.5
IR νmax 3392 (NH) and 1083 (S=O). MS (EI) m/z 254 (M+).
eq.) in 25 mL of H2O/AcOH (4:1) were stirred at 25°C for 22
HRMS (EI) calcd for C11H14N2OS2 (M+) m/z 254.0548.
h and gave 11 (1.01 g, 90%) as a white powder. Mp: 226-
Found: 254.0560. 1HNMR: δ0.99 (3H, t, J= 7.34,
227°C. IR νmax 3352 (NH), 1731 (C=O), 1276 and 1131. MS
CH3CH2CH2SO), 1.47- 1.79 (2H, m, CH2CH2SO), 2.75 (3H,
(EI) (m/z): 297 (M+). H-RMS (EI) calcd for C12H15N3O4S
s, CH3S), 2.68-2.87 (2H, m, CH2SO), 7.38 (1H, dd, J= 8.4, J=
(M+) 297.0783. Found: 297.0792. 1HNMR: δ 0.89 (3H, t, J =
1.5, H-6), 7.58 (1H, dd, J = 8.4, J = 0.6, H-7), and 7.69 (1H,
7.5, CH3CH2CH2SO2), 1.55 (2H, sext, J = 7.5, CH2CH2SO2),
dd, J = 1.5, J = 0.6, H-4), and 13.2 (bs, NH, int. D2O).
3.22 (2H, t, J = 7.5, CH2SO2), 3.80 (3H, s, CH3O), 7.58 (1H,dd, J = 8.2; J = 1.5, H-6); 7.62 (1H, d, J = 8.2, H-7), 7.91 (1H,
5-Chloro-6-(2,3-dichlorophenoxy)-2-(methylsulfinyl)-1H-
s, H-4); and 12.06 (bs, NH, int. D2O). HPLC: rt: 5.21 min. benzimidazole (8). To a stirred solution of 3 (0.50 g, 1.37 mmol) in 50 mL of CHCl3 was slowly added, dropwise, a Methyl [5-(phenylsulfinyl)-1H-benzimidazol-2-yl]carba-
solution of MCPBA (0.338g, 1.37 mmol) in 4 mL of CHCl3 at
mate (6). Following the general procedure, 2 (0.5 g, 1.67
0-5°C. The progress of the reaction was monitored by TLC
mmol) in 13 mL of AcOH/CH3CN and NaIO4 (0.393 g, 1.84
(CHCl3-MeOH, 95.5:0.5). At the end of the reaction the sol-
mmol) in 6 mL of H2O/AcOH (5:2) were stirred at 15ºC for 2
vent was removed in vacuo without heating, the residue was
h and gave 6 (0.527 g, 95%) as a pale pink solid, after recrys-
suspended in brine and neutralized with a saturated solution of
tallization from CHCl3. Mp 253.9ºC. TLC (Toluene-THF-
potassium carbonate. The mixture was extracted with CH2Cl2
AcOH, 5:1:1). IR νmax 3388 (NH), 1721 (C=O), 1047. MS
(3x20 mL). The combined organic extracts were dried with
(EI) (m/z): 315 (M+). HR-MS (EI) calcd for C20H13N3O3S
anhydrous sodium sulphate, filtered and evaporated in vacuo
(M+) m/z: 315.0678. Found: 315.0677. 1HNMR δ 3.76 (3H, s,
to give 8 (0.389 g, 75%) of a white soapy powder. Mp: 176-
CH3O), 7.36 (1H, dd, J = 8.4; J= 1.35, H-6), 7.4-7.55 (5H, m,
178°C. IR νmax: 3168 (NH), 1050 (SO). MS (EI) (m/z): 376
H-2’, H-3’, H4’, H5’, H-6’), 7.66 (1H, d, J = 8.4, H-7), 7.73
(M+). HRMS (EI) Calcd for C14H9Cl3N2O2S (M+) m/z
(1H, d, J = 1.35, H-4), and 11.86 (s, NH, int. D2O).
375.9450. Found: 375. 9422. 1HNMR: δ3.08 (3H, s, CH3SO),6.75 (1H, d, J = 8.4, H-6’), 7.28 (1H, t, J = 8.0, J = 8.4, H-5’),
Methyl [5-(phenylsulfonyl)-1H-benzimidazol-2-yl]carba-
7.40 (1H, dd, J = 8.0, J = 0.8, H-4’), 7.47 (1H, s, H-7), 7.93
mate (7). Following the general procedure, 2 (0.16 g, 0.54
(1H, s, H-4), and 13.82 (bs, NH, int. D2O).
mmol) in 15 mL of AcOH/CH3CN and NaIO4 (0.285 g, 1.34mmol) in 4.5 mL of H2O/AcOH (4:1) were stirred at 60°C for
5-Chloro-2-(methylsulfinyl)-5-(1-naphtyloxy)-1H-benzimi-
24 h and gave 7 (0.113 g, 67%) as a pale pink solid. Mp: dazole (13). Into a stirred solution of 12 (0.50 g, 1.476 mmol)
319.8-321.1°C. TLC (Toluene-THF-AcOH, 5:1:1). IR νmax
in 20 mL of CHCl3 was slowly added, dropwise, a solution of
3342 (NH), 1731 (C=O), 1268, and 1047 (SO2). MS (FAB)
MCPBA (0.394 g, 1.37 mmol) in 15 mL of CHCl3 at 0-5°C.
(m/z): 332 (M+1), HR-MS calcd for C15H14N3O4S (M+) m/z:
The progress of the reaction was monitored by TLC (CHCl3-
331.0627, found: 332.0726. 1H-NMR: δ 3.74 (3H, s, CH3O),
MeOH, 97:3). When the reaction was completed, it was treat-
7.36 (1H, dd, J = 8.4, J = 1.8, H-6), 7.48-7.53 (3H, m, H-3’,
ed with a solution of NaHCO3 until pH 7. Afterwards, the
H-4’, H-5’), 7.49 (1H, dd, J = 8.4, J = 0.6, H-7), 7.73 (1H, d, J
mixture was extracted with CHCl3 (3 × 3 mL). The combined
= 1.8, H-4), and 11.81 (s, NH, int. D2O).
organic extracts were dried with anhydrous sodium sulphate, filtered and evaporated in vacuo to give 13 as a white soapy 5-(Propylsulfinyl)-2-(trifluoromethyl)-1H-benzimidazole
powder. The solid was recrystallized from ethanol-benzene
(11). Following the general procedure, 10 (0.40 g, 1.54 mmol)
1:1 to give 0.381 g (72.43%) of a white powder. Mp 189-
in 8 mL of AcOH/CH3CN and NaIO4 (0.328 g, 1.69 mmol,
190°C. IR νmax 3422 (NH), 1048 (SO). MS (EI) (m/z): 356
1.09 eq.) in 16 mL of H2O/AcOH (5:2) were stirred at 60°C
(M+). HRMS (EI) Calcd for C18H13ClN2O2S (M+) m/z
for 2 h and gave 11 (0.381 g, 90%) as a white powder, after
356.0386 Found: 356.0380 1H NMR: δ 3.08 (3H, s, CH3SO),
recrystallization from cyclohexane-toluene. Mp: 123.2-125.2
6.702 (1H, d, J = 7.5, H-2’), 7.376 (1H, s, H-4), 7.403 (1H, t,
°C. IR νmax 3425 (NH), 1015 (SO). MS (m/z): 276 (M+), HR-
J = 8.1, H-3’), 7.568-7.638 (2H, m, H-6’, H-7’), 7.69 (1H, d,
Studies on the Selective S-oxidation of Albendazole, Fenbendazole, Triclabendazole, and Other Benzimidazole Sulfides
J = 8.4, H-4’), 7.97 (1H, s, H-7), 7.99-8.00 (1H, m, H-5’),
Computational methodology
8.18-8.21 (1H, m, H-8’), and 14.12 (bs, NH, int. D2O).
Complete optimization of the geometry of compound 17 was 4-(Propylthio)-o-phenylenediamine (15). A stirred mixture
done with the program Spartan’02 [21] at level RHF/6-
of 14 (0.5 g, 2.35 mmol), SnCl2·2H2O (3.18 g, 14.13 mmol)
31G(d,p). The electrostatic potential map was calculated from
and 5 mL of absolute ethanol was heated at 80°C under N2 for
2 h. The progress of the reaction was monitored by TLC(CHCl3-MeOH, 95.5:0.5), and once finished, it was allowed toreach room temperature, then, it was neutralized with a 50%
Acknowledgements
NaOH solution and filtered. The residue of tin salts was driedunder vacuum and extracted with AcOEt (3x10 mL). The
We are grateful to the Departamento Sistemas Biológicos
combined organic extracts were washed with brine, and dried
from the UAM-X for the financial support for this work and to
with anhydrous Na2SO4. After evaporation of the solvent
DGAPA, UNAM, for financing project IN 202101. We are
under vacuum, a brown viscous liquid was obtained. The
also grateful to Rosa Isela del Villar, Georgina Duarte,
crude product was immediately used in the next reaction with-
Margarita Guzmán and Marisela Gutiérrez, from the Facultad
de Química, UNAM, for the determination of the spectra. 5-(Propylthio)-1H-benzimidazole-2-thiol (16). A stirred mixture of 15 (0.429 g, 1.912 mmol), EtOH (6 mL), KOH References
(0.233 g, 3.53 mmol) in water (1 mL) and CS2 (0.2 mL, 3.532mmol) was heated at 50°C under N2 for 3 h. Then, the reaction
1. Gyurik, R. J.; Chow, A. W.; Zaber, B.; Brunner, E. L.; Miller, J.
was left 12 h at room temperature. The progress of the reac-
A.; Villani, A. J.; Petka, L. A.; Parish, R. C. Drug MetabolismDisposition. 1981, 9, 503-508.
yellow precipitate formed was poured into water and treated
2. Averkin, E. A.; Beard, C. C.; Dvorak, C. A.; Edwards, J. A.;
Fried, J. H.; Kilian, J. G.; Schiltz, R. A. J. Med. Chem. 1975, 18,
with 20% AcOH solution to pH 6. The solid was separated by
filtration, washed with water and air dried to obtain 16 (0.391
3. Lecaillon, J. B.; Godbillon, J.; Campestrini, J.; Naquira, C.;
g, 74%) of a slightly yellow powder. Mp: 216.1-217.8°C. IR
Miranda, L.; Pacheco, R.; Mull, R.; Poltera, A. A. J. Clin.Pharmacol. 1998, 45, 601-604.
max 3439 (NH). MS (m/z): 224 (M+). HRMS (EI) Calcd for
4. Alvarez, L. I.; Sánchez, S. F.; Lanusse, C. E. J. Vet. Pharmacol.
10H12Cl3N2O2S (M+) m/z 224.0442, found: 375. 9422. 1H-
Therap. 1999, 22, 77-86.
NMR: δ 0.99 (3H, t, J = 7.28, CH3CH2CH2S), 1.60 (2H, sext,
5. Virkel, G.; Lifschitz, A.; Soraci, A.; Sansinanea, A.; Lanusse, C. J = 7.28, CH2CH2S), 2.84 (2H, t, J = 7.28, CH2S), 7.14 (1H, s,
Xenobiotica 2000, 30, 381-393.
H-7), 7.15 (1H, s, H-6), 7.25 (1H, m, H-4), and 9.55 (bs, NH,
6. Murray, M.; Hudson, A. M.; Yassa, V. Chem. Res. Toxicol. 1992,
7. Szprengier-Juszkiewicz, T.; Semeniuk, S.; Wlodarczyk, B. Bull.Vet. Inst. Pulawy. 2002, 46, 119-125. 2-(Methylthio)-5-(propylthio)-1H-benzimidazole (17). Into a
8. Sanyal, P. K. Indian J. Pharmacol. 1994, 26, 200-203.
stirred, dark solution, of 16 (1.2 g, 5.33 mmol) in 4.5 mL of
9. Takeba, K.; Fujinuma, K.; Sakamoto, M.; Miyazaki, T.; Oka, H.;
acetone and KOH (0.351 g, 6.27 mmol) in 0.5 mL of water,
Itoh, Y.; Nakazawa, H. J. Chromatog. A. 2000, 882, 99-107.
10. De Laurentis, N.; Milillo, M. A.; Bruno, S. Pharm. Pharmacol.
2, CH3I (0.4 mL, 5.33 mmol) at 0°C.
Then, the mixture was stirred for 30 min at 10°C. The progress
Lett. 1996, 6, 51-53.
11. Xie, J.H.; Hu, Y.Z. Xhejiang Daxue Xuebao Yixueban. 31, 45
of the reaction was monitored by TLC (CHCl3-MeOH,
(2002); Chem. Abstr. 138, 73201(2002).
95.5:0.5) and once finished it was neutralized with a 20% HCl
12. Brandon, D. L.; Binder, R. G.; Bates, A. H.; Montangue, W. C. J.
solution and concentrated under vacuum. The residue was
Agric. Food Chem. 1994, 42, 1588-1594.
taken up with AcOEt, the extract washed with brine, dried with
13. Iddon, B.; Kutschy, P.; Robinson, A.G.; Suschitzky, H.; Kramer,
W.; Neugebauer, F.A. J. Chem. Soc. Perkin Trans. 1. 1992,
2SO4 and half concentrated under vacuum.
Addition of MeOH allowed the formation of 14 (1.22 g, 96%)
14. Hudlicky, M. Oxidation in Organic Chemistry. ACS Monograph
of a white powder. Mp: 142.7-142.9°C. IR νmax 3426 (NH).
186. American Chemical Society. Washington D.C. 1990, 252.
MS (EI) m/z: 238 (M+). HRMS (EI) Calcd for C11H14N2S2 (M+)
15. Leonard, N. J.; Johnson, C. R. J. Org. Chem. 1961, 27, 282-284. m/z 238.0598, found: 238.0582. 1HNMR: δ 0.97 (3H, t, J = 7.5,
16. Hiskey, R. G.; Harpold, M. A. J. Org. Chem. 1967, 32, 3191-
3CH2CH2S), 1.61 (2H, sext., J = 7.5 Hz, CH3CH2CH2S),
17. Evans, B.J.; Doi, T.; Musker, W. K. J. Org. Chem. 1990, 55,
2.85 (3H, s, CH3S), 2.85 (2H, t, J = 7.5, CH2SO), 7.24 (1H, dd,
J = 8.4, J = 1.5, H-6), 7.42 (1H, d, J = 8.4, H-7), 7.55 (1H, d, J
18. Hay, M. P.; Wilson, W. R.; Denny, W A. Tetrahedron 2000, 56,
= 1.2, H-4), and 10.21 (bs, NH, int. D2O). J. Mex. Chem. Soc. 2005, 49(4)
19. Hernández-Campos, A.; Ibarra-Velarde, F.; Vera-Montenegro,
Adams, T. R.; Ochsenfeld, C.; Gilbert, A. T. B.; Kedziora, G. S.;
Y.; Rivera-Fernández, N.; Castillo, R. Chem. Pharm. Bull. 2002,
Rassolov, V. A.; Maurice, D. R.; Nair, N.; Shao, Y.; Besley, N.
A.; Maslen, P. E.; Dombroski, J. P.; Daschel, H.; Zhang, W.;
20. Navarrete-Vázquez, G.; Yépez, L.; Hernández-Campos, A.;
Korambath, P. P.; Baker, J.; Byrd, E. F. C.; Van Vooris, T.;
Tapia, A.; Hernández-Luis, F.; Cedillo, R.; González, J.;
Oumi, M.; Hirata, S.; Hsu, C.-P.; Ishikawa, N.; Florian, J.;
Martínez-Fernández, A.; Martínez-Grueiro, M.; Castillo, R.
Warshel, A.; Johnson, B. G.; Gill, P. M. W., Head-Gordon, M.;
Bioorg. Med. Chem., 2003, 11, 4615-4622.
Pople, J. A. J. Comput. Chem. 2000, 21, 1532-1548.
21. Kong, J.; White, C. A.; Krylov, A. I.; Sherrill, C. D.; Adamson,
R. D.; Furlani, T. R.; Lee, M. S.; Lee, A. M.; Gwaltney, S. R.;
SIGMA-ALDRICH sigma-aldrich.com Fiche de Données de Sécurité / Fiche Signalétique 1. IDENTIFICATION DU PRODUIT ET DE LA SOCIETE : Pour des fins de recherche en laboratoire. (Pour le fournisseur et le fabricant) Renseignements sur la Product Safety - Americas Region 1-800-521-8956 2. IDENTIFICATION DES DANGERS Aperçu des urgences Organes cibles WHMIS Class
International Journal of Pharmacy Teaching & Practices 2012, Vol.3, Issue 4, 429-433. Role of Bisoprolol Adding oo Ace Inhibitor and Fu rosemide Combination on the Left Ventr icular Function in Systolic Heart Failure Patients Efta T riastuti 1*, Dadang Hendrawan2, Muha mmad Saifurrohman 3 1Study Program of Pharmacy, Faculty of Medicine, Braw ijaya University, Malang

 Studies on the Selective S-oxidation of Albendazole, Fenbendazole, Triclabendazole, and Other Benzimidazole Sulfides
Scheme 1. Reagents: (a) SnCl2·2H2O, EtOH; (b) CS2, KOH, EtOH; (c) CH3I, KOH, CH3COCH3; (d) NaIO4.
Studies on the Selective S-oxidation of Albendazole, Fenbendazole, Triclabendazole, and Other Benzimidazole Sulfides
Scheme 1. Reagents: (a) SnCl2·2H2O, EtOH; (b) CS2, KOH, EtOH; (c) CH3I, KOH, CH3COCH3; (d) NaIO4.