Tadalafil gehört zur Gruppe der PDE5-Hemmer und wirkt über eine hochselektive Blockade des Enzyms Phosphodiesterase Typ 5. Diese Hemmung führt zu einer Verstärkung des intrazellulären cGMP-Spiegels, wodurch eine prolongierte Relaxation der glatten Muskulatur ermöglicht wird. Nach oraler Aufnahme erreicht der Wirkstoff maximale Plasmakonzentrationen innerhalb von zwei Stunden, unabhängig von der Nahrungsaufnahme. Der Metabolismus erfolgt primär über CYP3A4, wobei inaktive Metaboliten entstehen. Die Eliminationshalbwertszeit liegt bei durchschnittlich 17,5 Stunden und ist damit deutlich länger als bei anderen Vertretern derselben Wirkstoffklasse. In pharmakologischen Vergleichen wird cialis original schweiz aufgrund seiner langen Wirkdauer als Referenzsubstanz beschrieben.
Doi:10.1016/j.ijpharm.2005.11.025
International Journal of Pharmaceutics 309 (2006) 199–207
1. Properties and in vitro/in vivo behavior of acyclovir
Yiguang Jin , Li Tong , Ping Ai , Miao Li , Xinpu Hou
a Department of Pharmaceutical Chemistry, Beijing Institute of Radiation Medicine, Beijing 100850, PR China
b Department of Physical Pharmacy, School of Pharmaceutical Sciences, Peking University, Beijing 100083, PR China
c Department of Biochemistry and Biotechnology, College of Life Sciences, Beijing Normal University, Beijing 100875, PR China
Received 1 September 2005; received in revised form 8 November 2005; accepted 15 November 2005
Abstract
Self-assembled drug delivery systems (SADDS) were designed in the paper. They can be prepared from the amphiphilic conjugates of hydrophilic
drugs and lipids through self-assembling into small-scale aggregates in aqueous media. The outstanding characteristic of SADDS is that they arenearly wholly composed of amphiphilic prodrugs. The self-assembled nanoparticles (SAN) as one of SADDS had been prepared from the lipidderivative of acyclovir (SGSA) in the previous paper. They were further studied on the properties and the in vitro/in vivo behavior in this paper. The SAN kept the physical state stable upon centrifugation or some additives including some inorganic salts, alkaline solutions, surfactants andliposomes except for HCl solution, CaCl2 solution and animal plasma. Autoclave and bath heat for sterilization hardly influenced the SAN. However,gamma-irradiation strongly destroyed the structure of SAN and SGSA was degraded. SGSA in SAN showed good stability in weak acidic or neutralbuffers although it was very sensitive to alkaline solutions and carboxylester enzymes, the half-lives (t1/2) of which in the buffer at pH 7.4, thealkaline solution at pH 12.0, pig liver carboxylester enzyme solution, rabbit plasma, and rabbit liver tissue homogenate were 495, 21, 4.7, 25 and8.7 h, respectively. Compared with SGSA in a disordered state, the specific bilayer structures of SAN could protect SGSA from hydrolysis throughhiding the sensitive ester bonds. The SAN showed hemolytic action because the amphiphilic SGSA could insert into rabbit erythrocyte membranes. Both the high concentration of SGSA in samples and the long incubation time improved hemolysis. No hemolysis was observed if the additionalvolume of the SAN was less than 10% of rabbit whole blood in spite of the high concentration of SGSA. Plasma proteins could interfere theinteraction between the SAN and erythrocytes by binding the SAN. The in vitro antiviral activity of acyclovir SAN was limited possibly becauseof the weak hydrolysis of SGSA in Vero cells, and the SAN showed a little cell toxicity possible due to the amphiphilicity of SGSA. A macrophagecell line of QXMSC1 cells showed uptake of the SAN but not significantly. The SAN were rapidly removed from blood circulation after bolus ivadministration to rabbits with the very short distribution t1/2 (1.5 min) and the elimination t1/2 (47 min). The SAN were mainly distributed in liver,spleen and lung after iv administration, and SGSA was eliminated slowly in these tissues (t1/2, about 7 h). It would appear that the nanosized SANwere trapped by the mononuclear phagocyte system. SADDS including SAN combine prodrugs, molecular self-assembly with nanotechnology,and hopefully become novel drug delivery approaches. 2005 Elsevier B.V. All rights reserved. Keywords: Prodrugs; Molecular self-assembly; Nanotechnology; Amphiphiles; Acyclovir; Mononuclear phagocyte system
1. Introduction
possess two elements: the ability to target and drug controlledrelease. Drug targeting will ensure high therapeutic efficacy.
The efficacy and safety of drugs is always the key of phar-
But maybe even more important it will reduce side effects. The
macotherapy. To achieve the purpose, a variety of methods are
reduction or even prevention of side effects can also be achieved
applied to drug delivery. An ideal drug delivery system should
by controlled release. Drug carriers such as particulates (lipo-somes, nanoparticles, microemulsions) and externally triggered(pH-, temperature-, or magnetic-sensitive) carriers have widely
∗ Corresponding author. Tel.: +86 10 66931220; fax: +86 10 68214653. E-mail address: jin [email protected] (Y. Jin).
ever, almost all of current delivery systems load drugs passively
0378-5173/$ – see front matter 2005 Elsevier B.V. All rights reserved. doi:10.1016/j.ijpharm.2005.11.025
Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
so that the problems of low drug-loaded efficiency and drug leak-
Acyclovir is a typical nucleoside antiviral agent against her-
age in preparation, preservation and transport in vivo always
pes simplex virus (HSV) with the low oral bioavailability (20%)
and a short circulation half-life (t1/2, 2.5 h)
ers could have been destroyed in vivo before reaching target
Some lipophilic derivatives of acyclovir showed
sites. In addition, lipophilic biomembranes including cell mem-
branes usually prevent hydrophilic drugs from entering into
acyclovir as a model drug to study SADDS in this paper. A
target sites (Generally, carriers also can-
series of long-chained lipid derivatives of acyclovir had been
not override cell membranes unless endocytosis/phagocytosis
synthesized and the self-assembly of them were characterized
by cells/macrophages, so that the loaded drugs are probably
released on target surfaces. A majority of drugs could not reach
stearyl-glycero-succinyl-acyclovir (SGSA), the single-chained
target sites at all on account of the poor properties of carriers
lipid derivative self-assembled into cuboid-like self-assembled
nanoparticles (SAN) in water based on the hydrophobic interac-
It is well known that the surfactant-like amphiphiles would
tion of lipid chains and the layer-by-layer hydrogen bonding
like to self-assemble into ordered aggregates such as micelles,
of nucleoside moieties. Acyclovir SAN had an average size
vesicles, hexagosomes and cubosomes. Some of the aggregates
of 83.2 nm, a negative surface charge of −31.3 mV. It should
be rationally regarded as one of SADDS. The properties and
the in vitro/in vivo behavior of acyclovir SAN were investi-
Unfortunately, surface-active drugs espe-
gated in the paper, mainly including stability, the interaction
with cells, pharmacokinetics and tissue distribution after iv
However, some amphiphiles with potential pharmacological
action (drugs or prodrugs) can be prepared, and they likely self-assemble into ordered aggregates. Optimally, the aggregates can
2. Materials and methods
deliver themselves in vivo. We name the aggregates as self-assembled drug delivery systems (SADDS). Based on the aim
of drug delivery, SADDS had better be small-scale systems andthey would show drug targeting and sustained release. But the
The lipid derivative of acyclovir, SGSA was prepared accord-
outstanding characteristic of SADDS over common nanopar-
ing to the previous research (Analytical reagents
ticles or liposomes is that they are nearly wholly composed of
were used, and chromatographic reagents were used in HPLC
amphiphilic prodrugs, so that high drug-loaded amount and very
determination. Distilled water was always used. Surfactants used
low drug leakage are archived easily. In addition, the amphiphilic
in the stability investigation of SAN were from Amresco (sodium
monomers of SADDS would like to permeate biomembranes of
dodecyl sulphate, SDS), Sigma (Brij 35), Beijing Chemical
targets provided that SADDS were decomposed on target sur-
Reagents Company (Tween 80 and cetyltrimethylammonium
bromide, CTAB), Shenyang Yaoda Jiqi Pharmaceutical Fac-
Vaizoglu and Speiser used the word ‘pharmacosomes’ to
tory (poloxamer 188) and Lucas Meyer GmbH (soybean phos-
describe the colloidal dispersions prepared from drug-lipid con-
phatidylcholine, SPC). Liposomes were prepared from SPC by
jugates with or without additional surfactants (
Pindolol pharmacosomes (vesicle-like) were
prepared from pindolol diglyceride by the authors. Pharma-
Pig liver carboxylester enzyme (PLCE, Sigma) was dissolved
cosomes have been not deeply studied, possibly because no
in Tris–HCl buffer (20 mM, pH 7.4) before use. A macrophage
appropriated theory supports the new dosage form and no appro-
cell line of QXMSC1 cells was from Department of Biotechnol-
priated drugs and lipids are selected. Obviously, pharmacosomes
ogy, Beijing Institute of Radiation Medicine (BIRM). African
can be considered as one of SADDS based on the theory of the
green monkey kidney (Vero) cells were from Institute of Medic-
inal Biotechnology, Chinese Academy of Medical Sciences.
The structural modification of drugs is necessary to obtain
Male albino rabbits (1.9–2.6 kg) from Laboratory Animal
amphiphilicity. The lipid derivation of hydrophilic drugs should
Center of BIRM were used. Principles in good laboratory animal
be preferentially considered because lipid derivatives can be well
care were followed and animal experimentation was in compli-
degraded in vivo and permeate biomembranes (
ance with the Guidelines for the Care and Use of Laboratory
Nucleoside antivirals have to be activated to phosphates in cell
Animals in BIRM. The rabbits were sacrificed by euthanasia
plasma to resist virus although many of them permeate into cells
to remove tissues. The homogenates used in the experiments
not well due to high molecular polarity (
of chemical stability and tissue distribution were prepared in
Some lipophilic derivatives of nucleosides self-assemble
in organic solvents based on hydrogen bonding and some phospholipid-
nucleoside conjugates form ordered aggregates in aqueous solu-tions (
Acyclovir SAN were prepared with a controlled process
Therefore, nucleoside antivirals are the optimal precursors to
according to the previous research (The prepara-
prepare self-assembling amphiphilic prodrugs.
tion method is described as follows. SGSA solutions of 5 mg/ml
Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
in tetrahydrofuran (THF) were slowly and continually injected
additional volume of additives was, respectively, 20, 50, 100 and
into vortexed water under surface via a 100-l micro-syringe
until a homogeneous and slightly opalescent suspension wasobtained. The suspensions were incubated in a 37 ◦C water bath
under vacuum for removing solvents and being concentrated.
Acyclovir SAN were diluted with different buffers, includ-
The final suspensions could contain high-concentration SGSA
ing 20 mM phosphate buffers (pH 5.0 and 7.4) and 20 mM
of more than 15 mg/ml, and appeared homogeneously.
Tris–HCl buffers (pH 9.0 and 12.0), and the dilutions were incu-bated in a 37 ◦C bath. At predetermined time intervals, 20-l
2.3. HPLC determination of acyclovir and its derivatives
aliquots were removed, dissolved with methanol, and assayedby HPLC. SGSA solutions in dimethylsulphoxide (DMSO) were
High-performance liquid chromatographic (HPLC) experi-
also mixed with buffers, and the stability was measured to com-
ments were performed at room temperature on a Shimadzu
pare the degradation kinetics with SGSA in SAN.
10A HPLC system (Japan) that consisted of LC-10Avp pump,
The effects of PLCE solution (10 U/ml), rabbit plasma and
SPD-10Avp UV detector, SCL-10Avp controller, and Shi-
tissue homogenates on the chemical stability of acyclovir SAN
madzu CLASS-VP 6.02 chromatographic workstation soft-
at 37 ◦C were investigated as above. The samples were depro-
ware. The DiamonsilTM C18-ODS HPLC columns (5 m,
teinized with methanol, followed by vortex for 1 min, and cen-
250 mm × 4.6 mm) and the EasyGuardTM C18-ODS HPLC
trifuged at 10,000 rpm for 10 min. SGSA in supernatants was
guard columns (5 m, 8 mm × 4 mm) were purchased from
determined by HPLC. Acyclovir in samples was also deter-
Dikma (China). A manual injection valve and a 20-l loop
mined as the above procedure except for deproteinization with
(7725i, Rheodyne, USA) were used. UV detector was fixed at
Since the polarity of acyclovir, succinyl-acyclovir (SACV,
the synthesis intermediate or the possible hydrolysis product)
Gamma-irradiation of 1.5 × 104 Gy from a 60Co source
and SGSA was significantly different, the various mobile phases
(BIRM, China) was used to sterilize acyclovir SAN in glass
were used for their HPLC determination. Acyclovir and SACV
bottles at room temperature. The heat sterilization of the SAN
in all samples except for tissue homogenates were determined
in glass bottles was performed through autoclave for 30 min, or
in the mobile phase of methanol/water (19:81, v/v) containing
the 100 ◦C bath for 30 min. Whether SGSA in the sterilized SAN
40 mM ammonium acetate (pH 7.0) at 0.8 ml/min. The reten-
was hydrolyzed was evaluated by HPLC determination.
tion times (tR) of acyclovir and SACV were about 6.8 and7.6 min, respectively. The mobile phase used to determine acy-
2.5. Interaction between SAN and erythrocytes
clovir in tissue homogenates was methanol/water (12:88, v/v)containing 40 mM ammonium acetate (pH 7.0), and the flow
Rabbit erythrocyte suspension (2%, v/v) was prepared as fol-
rate was 1.0 ml/min. The tR of acyclovir was about 8.7 min.
lows. Whole blood was obtained from rabbits via marginal ear
SGSA in all samples was determined with the mobile phase
vein puncture and collected in a clean beaker, and agitated with
of methanol/water (80:20, v/v) containing 40 mM ammonium
a glass stick to remove fibrinogen. Erythrocytes were separated
acetate (pH 7.0) at 0.8 ml/min. The tR of SGSA was about
by centrifugation at 3000 rpm for 3 min, and washed three times
with 0.9% NaCl solution. The sediment cells were diluted with0.9% NaCl solution to obtain the erythrocyte suspension that
2.4. Stability investigation of SAN
was stored at 4 ◦C and used within 24 h.
Hemolytic action was investigated briefly as the followings.
The 2-ml samples containing 1% erythrocytes, a series of SAN
The turbidity method was often used to investigate the phys-
suspensions and supplementary 0.9% NaCl solutions were pre-
pared. After 12 h of incubation at 37 ◦C, hemolytic phenomena
were observed by naked eyes against the completely hemolytic
equal to the absorbance at 550 nm with water as references in
sample prepared by adding water into erythrocytes, and the cells
the paper. Particle size increasing or particle aggregation can be
were counted with light microscopy. The cells and the super-
expressed by turbidity increasing. The effects of centrifugation
natants were separated, and SGSA in them was determined by
and additives on the physical stability of the SAN were inves-
HPLC. Acyclovir solutions instead of SAN were as control.
tigated. The SAN were centrifuged at various rotate speeds for
The interaction between acyclovir SAN and rabbit whole blood
5 min and then resuspended thoroughly. The turbidity was mea-
(fresh heparinized) was also investigated.
sured by a Shimadzu UV-2501PC spectrophotometer. The waterdiluted SAN (containing about 120 g/ml SGSA) of 2.5 ml were
2.6. Toxicity and antiviral activity on cell model
added into cuvettes, followed by, respectively, adding with avariety of additives, agitated thoroughly, and then the turbid-
Vero cells were cultured at 37 ◦C in a humidified atmosphere
ity was measured. The additives included 150 mM NaCl, 0.1 M
of 5% CO2. The cultural medium was the MEM (Sigma) solution
NaOH, 100 mM SDS, 10 mM CTAB, 10% Tween 80, 10% Brij
supplemented with 10% calf serum and 1 × 105 unit penicillin
35, 10% poloxamer 188, SPC liposomes and rabbit plasma. The
and streptomycin. Vero cells were cultured in 96-well plates
Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
at a concentration of 2 × 104 cells/well. After 24 h of culture,
tissues were removed, weighted and disrupted to homogenates.
the autoclaved SAN suspensions and acyclovir solutions diluted
The plasma samples and the homogenate samples were stored
with the cultural medium not containing serum were, respec-
at −20 ◦C until HPLC analysis (see Section
tively, added to the wells. Eight samples of the SAN with SGSAconcentration increasing from 31.2 to 4000 g/ml were added
3. Results and discussion
to the wells in triplicates. So did eight samples of acyclovir solu-tions from 7.8 to 1000 g/ml. The wells not containing the drugs
were as control. After 48 h of incubation at 37 ◦C, the cytopathiceffect (CPE) assay was performed with light microscope, and the
Like other colloidal dispersions, SAN may aggregate into
large particles. After centrifugation with rotate speed from 2000
Anti-HSV therapeutic effect was also investigated like the
to 8000 rpm, the particle size of acyclovir SAN increased slightly
above procedure. After Vero cells were cultured in 96-well plates
with speed dependency according to the turbidity measurement
for 24 h, they were infected by herpes simplex virus type-1
(data not shown). However, the particles size increased sig-
(HSV-1) strain (VR733, ATCC, USA) of 104-fold dilution. The
nificantly after 10,000-rpm centrifugation. The surface charge
cultural media were withdrawn after 2 h of virus adsorption.
repulsion among SAN improves to keep stable (
Eight samples of the SAN with SGSA concentration increasing
In fact, nearly no significant precipitants or flocculation
from 1.9 to 250 g/ml were added to the wells in triplicates. So
appeared after the SAN containing SGSA of less than 5 mg/ml
did eight samples of acyclovir solutions from 0.8 to 100 g/ml.
kept at room temperature for one year.
The wells without HSV-1 infection were as control. After 48 hof incubation at 37 ◦C, the CPE was examined, and the 50%
The low-concentration SAN would like to keep stable when
being mixed with 150 mM NaCl, 0.1 M NaOH, 100 mM SDS,10 mM CTAB, 10% Tween 80, 10% Brij 35, 10% poloxamer
2.7. Macrophage culture and uptake of SAN
188 and SPC liposomes because the turbidity of the mixtures hadno significant changes, even though the large volume (500 l)
A macrophage cell line of QXMSC1 cells in 6-well plates
of additives was used to mix with the SAN of 2.5 ml. How-
was cultured at 37 ◦C in a humidified atmosphere of 5% CO2.
ever, 0.1 M HCl solution, 150 mM CaCl2 solution and plasma
The cultural medium was the RPMI-1640 (Sigma) solution sup-
significantly increased the turbidity even when a few additives
plemented with 10% calf serum, 2.0 mM glutamine, 0.05 mM
(50 l) were added. H+ and Ca2+ could promote the negatively
2-mercaptoethanol, 4.5 g/l glucose, 1.5 g/l NaHCO3, 1.0 mM
charged SAN aggregating or fusion by possibly improving sur-
sodium pyruvate, 10 mM HEPES, and 1 × 105 unit penicillin
plasma on SAN could mainly be resulted from plasma protein
1 × 105 cells/well, the autoclaved SAN diluted with the cul-
binding to the SAN, and the protein could improve the aggrega-
tural medium not containing serum, were added to each well.
After the plates had been incubated at 37 ◦C for a predeter-
the sensitivity of SAN to NaCl-like electrolytes was depended on
mined period, the supernatants in the wells were drawn off, and
the particle number per unit volume of suspensions. The low-
the cells were washed with the cold Tris–HCl buffer (20 mM,
concentration SAN (less than 5 mg/ml) were not sensitive to
pH 7.4). The washing was collected and added to the super-
150 mM NaCl. However, when SGSA in SAN was over 7 mg/ml,
natants. The remaining cells were mixed with Tris–HCl buffer
i.e. high particle number per unit volume, 150 mM NaCl solution
(0.5 ml), scraped off, collected in a centrifuge tube, and expe-
rienced probe sonication in a 0 ◦C bath for 20 s to prepare cell
Although the SAN were relatively insensitive to various
lysates. SGSA in supernatants and cell lysates was determined.
surfactants and liposomes, these surfactants including phospho-
Acyclovir solutions were as control.
lipids did not benefit the preparation of SAN. They were apt tointerfere the formation of SAN. A lot of large particles appeared
2.8. SAN iv administration to rabbits
if the surfactants were co-dissolved with SGSA in THF andinjected into water. The surfactants could insert the bilayers of
Pharmacokinetics and tissue distribution were studied after
SAN on preparing so that the SAN could not be ready to form.
acyclovir SAN bolus iv administration to rabbits. SGSA in SANshould be concentrated to 15 mg/ml or more to reduce injec-
3.1.3. Effects of sterilization methods
tion volume. The SAN were autoclaved and determined before
As a sterilization method, 60Co gamma-irradiation strongly
use, and then administered to rabbits at SGSA dose of 30 mg/kg
facilitated the SAN aggregation and damage. A lot of floc-
through ear vein. Half of one milliliter of blood sample was col-
culation appeared after irradiation, and the content of SGSA
lected into heparinized centrifuge tubes at 0, 0.25, 0.5, 1, 1.5, 2, 3,
decreased. Like liposomes, gamma-irradiation may destroy the
4, 5, 8, 10, 15, 20, 30, 40, 50, 60, 90, 120, 180, 240 min after med-
bilayer structures of SAN and promote monomer degradation
ication. Plasma was separated by centrifugation at 3000 rpm for
10 min. The rabbits were sacrificed at 0.5, 2, 4 and 12 h, and the
fewer influences than radiation on bilayers (
Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
The autoclave or the 100 ◦C bath for sterilization neither influ-
Interestingly, after SGSA solutions in DMSO were diluted
enced the appearance of the SAN nor improved SGSA hydroly-
with buffers, SGSA degraded more rapidly than SGSA in SAN.
sis. Therefore, autoclave or 100 ◦C bath is the good sterilization
The t1/2 of SGSA in the DMSO/buffers mixtures were 495 h
at pH 5.0 (about 1/4 of SGSA in SAN) and 210 h at pH 7.4(less than 1/2 of SGSA in SAN), and disappeared completely
within 20 min at pH 12.0. The degradation of SGSA is mainlydetermined by the exposure probability of sensitive ester bonds
to water according to the above analysis. The sensitive bonds
SGSA in SAN was very sensitive to alkaline solutions (pH
of SGSA in SAN are hidden in bilayers. However, when SGSA
9.0 and 12.0), and kept stable in weak acidic buffers (pH 5.0)
solutions in DMSO were diluted with buffers, SGSA molecules
and neutral buffers (pH 7.4) (SGSA possesses the
fell into an irregular state where a lot of sensitive ester bonds
carboxylester structure and would like to be hydrolyzed with
maybe directly exposed to water and catalyzers. Therefore, it
is probably the specific ordered structures of SAN that protect
hydrolysis was acyclovir while the possible intermediate SACV
hardly appeared, so that the ester bond between succinyl andacyclovir moieties was more sensitive than the bond between
3.2.2. Effects of hydrolase, plasma and tissue homogenates
succinyl and glycerol moieties. The pseudo-first order kinetics
Hydrolases including carboxylester enzymes exist widely in
was used to describe SGSA degradation. The t
organism. PLCE, rabbit plasma and rabbit tissue homogenates
SAN at pH 5.0, 7.4, 9.0 and 12.0 were 1733, 495, 94 and 21 h,
were used to study the enzymatic hydrolysis of SGSA. SGSA
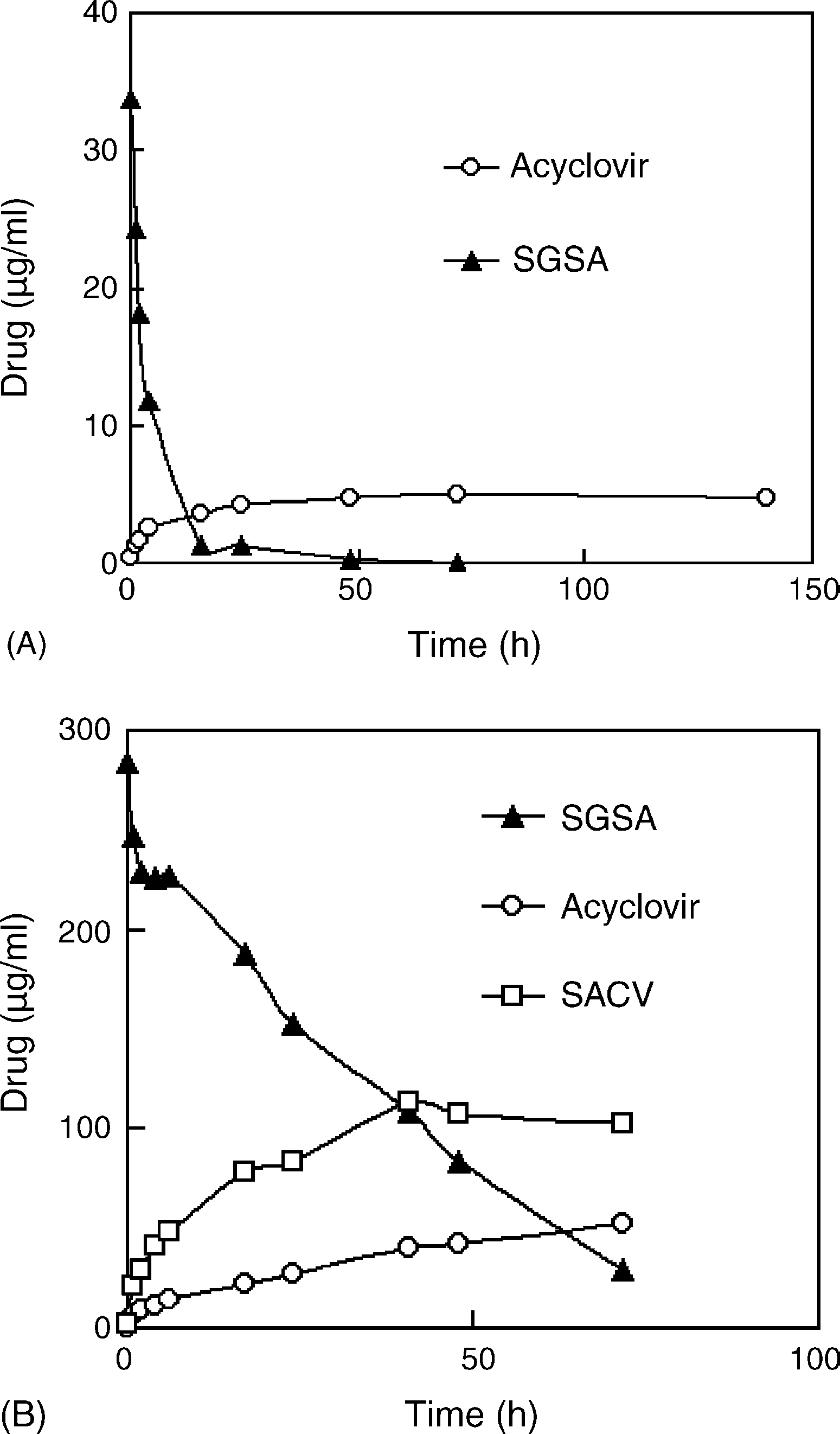
in SAN was nearly completely hydrolyzed in PLCE solutions(10 U/ml) after 15 h of incubation at 37 ◦C with the t1/2 of 4.7 h,and the hydrolysis speed was more than 100 folds as rapid asin pH 7.4 buffers (Therefore, SGSA is very sensi-tive to carboxylester enzymes. Also, rabbit plasma hydrolyzedSGSA much more rapidly than pH 7.4 buffers with the t1/2
Fig. 1. The pH dependency of the chemical stability of acyclovir SAN. TheSAN were diluted with different buffers and incubated at 37 ◦C. The bufferswere 20 mM phosphate buffers (pH 5.0 and 7.4) and 20 mM Tris–HCl buffers(pH 9.0 and 12.0), respectively. (A) The degradation profiles of SGSA; (B) the
Fig. 2. The effects of: (A) pig liver carboxylester enzyme (PLCE); and (B) rabbit
production profiles of acyclovir. The half-lives of SGSA at pH 5.0, 7.4, 9.0 and
plasma on the chemical stability of acyclovir SAN at 37 ◦C. PLCE solution
12.0 were 1733, 495, 94 and 21 h, respectively, based on the pseudo-first order
(10 U/ml) was prepared with Tris–HCl buffer (20 mM, pH 7.4) before use. The
half-lives of SGSA were 4.7 h in PLCE solution and 25 h in rabbit plasma. Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
of 25 h (Some intermediate SACV appeared in theplasma samples, which indicated that some enzymes in plasmacould hydrolyze the succinyl-glycerol ester bonds of SGSA. Thedegradation of SGSA in rabbit tissue homogenates depended onthe types of homogenates. The degradation t1/2 of SGSA in heart,liver, lung, spleen, kidney and brain were 5.0, 8.7, 11.8, 13.7,30.7 and 73.7 h, respectively, which must result from diverseenzyme activity between tissue homogenates.
Hemolytic phenomena partly appeared with varied extent
in the SAN/erythrocyte mixtures after 12 h of incubation at37 ◦C. Both the high-concentration SGSA and the long incuba-tion time improved hemolysis. SGSA of 18 g/ml did not show
Fig. 3. The uptake of acyclovir SAN by a macrophage cell line of QXMSC1cells after co-culturing them at 37 ◦C. Results were expressed as the mean ± S.D.
significantly hemolytic action, while marked hemolysis hap-
(n = 3). No significant uptake was found.
pened when SGSA in samples was over 36 g/ml. At the sametime, the cell number in the sample containing 18 g/ml SGSAwas 18 × 107, close to the primitive cell number (24 × 107),
not show any toxicity even when acyclovir was over 1000 g/ml,
while the sample containing 36 g/ml SGSA had 12 × 107 cells.
i.e. 4444 M. Referred to the interaction between SAN and ery-
When SGSA of 90 g/ml was in the sample, the cell num-
throcytes, the strong cell-membrane insertion of amphiphilic
ber was only 3.0 × 107. The concentration ratio of SGSA in
SGSA could be the primary reason of cell toxicity
cells and solutions (Ccell/Csolution) had positive linear relation-
ship with the cell number in samples, i.e. the more cell number
The anti-HSV IC50 of SGSA in SAN on Vero cell model
was, the more fraction SGSA distributed in cells. When enough
was 46.8 M, higher than that of acyclovir (7.1 M). Antiviral
SGSA molecules distributed in cells, cell membranes would
selection index (SI) was equal to TC50/IC50. The SI of SGSA and
like to be disrupted, and then SGSA together with hemoglobin
acyclovir were 24 and more than 626, respectively. The above
was released to solutions, which resulted in cell number and
data just demonstrate that the antiviral activity of SGSA in SAN
Ccell/Csolution reducing. The high-concentration SGSA in sam-
is much weaker than acyclovir, but whether the in vivo results
ples would accelerate and strengthen the process. In the case
of 90 g/ml SGSA, Ccell/Csolution were 1.2, compared with 3.3
Acyclovir has to be transformed to its triphosphate in cell
of 18 g/ml SGSA. Therefore, it is likely that the amphiphilic
plasma to resist HSV. The in vitro anti-HSV action of the SAN
SGSA molecules may insert into erythrocyte membranes until
demonstrated that SGSA could enter into cell plasma where
membrane breakdown like some surface-active chemicals
it was hydrolyzed to acyclovir. The degradation of SGSA in
Hemolysis did not immediately happen on mixing
organism was carboxylester enzyme-dependency (see Section
probably because the transfer of SGSA from SAN to erythrocyte
Unfortunately, the carboxylester enzyme activity of Vero
membranes needed time. Acyclovir solution did not show any
cells is low according to the literature (
hemolytic action. Cell-membrane insertion would give SGSA a
In fact, some prodrugs of antivirals show strong antiviral action
in vivo but weak activity on cell model because the enzyme
Surprisingly, SAN seemed to have no hemolytic effect on
activity in vivo and in vitro is significantly different (
whole blood. When the additional SAN was less 10% (v/v) of
For example, a good antiviral famciclovir (a
rabbit whole blood, no hemolysis happened even though SGSA
prodrug of acyclovir) do not show any anti-HSV action on Vero
in the sample was near to 1000 g/ml. However, as soon as the
additional SAN or water was over 10% (v/v) of whole blood,hemolysis preferred to happen. Obviously, the hemolysis ofwhole blood was just relevant to the descent of osmotic pres-
sure of samples. The binding plasma proteins would markedlyreduce the interaction between SAN and erythrocytes, and the
QXMSC1 cells are established from murine marrow and
binding action also named opsonization would lead the uptake
show phagocytic ability (The uptake of acy-
of SAN by the mononuclear phagocyte system (MPS) in vivo
clovir SAN by QXMSC1 cells was not significant (The
in vitro uptake of no-ligand modified liposomes by macrophages
is not significant too (However, afterbeing modified by ligands such as mannose residues and serum
3.4. In vitro toxicity and antiviral activity of SAN
poteins (opsonins), the liposomes or the nanoparticles can bemarkedly trapped by macrophages
Acyclovir SAN showed weak toxicity to Vero cells. TC50
of SGSA was 750 g/ml, i.e. 1126 M. Acyclovir solutions did
Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
Fig. 4. The time profile of SGSA concentration in plasma after acyclovir SANbolus iv administration to rabbits with 30 mg SGSA/kg through ear vein. Results
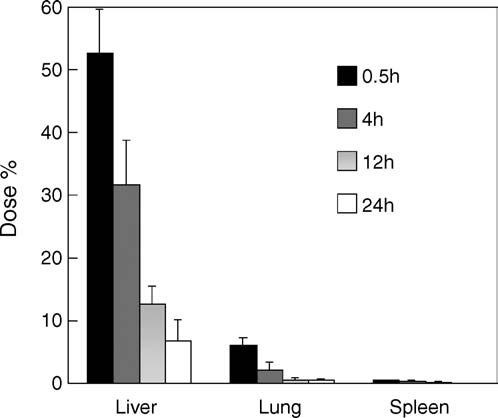
Fig. 5. The tissue distribution of SGSA after acyclovir SAN bolus iv admin-
were expressed as the mean ± S.D. (n = 5). The field of 0–5 min was considered
istration to rabbits. Results were expressed as the mean ± S.D. (n = 3). SGSA
as the distribution phase with the half-life (t1/2␣) of 1.5 min, and 5–90 min as
was mainly distributed in liver, spleen and lung although only a little SGSA
the elimination phase with the half-life (t1/2) of 47 min based on the first order
appeared in kidney and heart, and nothing was found in brain. The elimination
kinetics. A little higher SGSA concentration at 180 min than the minimum level
t1/2 of SGSA in liver, spleen and lung were 7.6, 6.8 and 6.6 h, respectively, near to
was maybe due to some lung-blocked SAN-aggregating large particles releasing
the hydrolysis t1/2 of SGSA in tissue homogenates. The main tissue elimination
way of SGSA could be resulted from the metabolism.
liver, spleen and lung were 7.6, 6.8 and 6.6 h, respectively, near
to the hydrolysis t1/2 of SGSA in tissue homogenates, so that
The SAN were cleared from blood circulation very quickly
the metabolism of SAGS in cell plasma could be the main elim-
after rabbits received acyclovir SAN by bolus iv administra-
ination way. From the plasma concentration-time curve
tion The plasma concentration of SGSA had descended
SGSA concentration in plasma at 180 min was a little higher than
for about 90% at 5 min, and approached the minimum level at
the minimum level, which was thought that some lung-blocked
30 min. The field of 0–5 min was considered as the distribution
SAN-aggregating large particles released SAN or SGSA to cir-
phase with the half-life (t1/2␣) of 1.5 min, and 5–90 min as the
culation again. Acyclovir as the in vivo metabolism product was
elimination phase with the half-life (t1/2) of 47 min based on
also determined, and no acyclovir was found in plasma, heart,
the first-order kinetics. The pharmacokinetic characteristics of
kidney and brain. Acyclovir with less than 10 g/g appeared in
SAN were similar to the particulate preparations such as lipo-
liver, spleen and lung within 12 h after administration because
acyclovir could be eliminated rapidly from plasma and tissues
(Section it was likely that the SAN were opsonized in vivo
The site-specific distribution of SAN indicates that, acyclovir
by plasma proteins, and then it would appear that the nanosized
SAN or other SAN prepared from antivirals can be expected to
SAN were trapped by the MPS. Some physicochemical proper-
benefit the therapy of virosis in liver, spleen and lung. The SAN
ties such as size, surface charge would also influence the in vivo
would be considered as the novel preparations of acyclovir with
clearance rate of the SAN. In addition, the clearance of the SAN
targeting and sustained-release functions although acyclovir was
seemed more rapid than liposomes or nanoparticles (
a model drug in the paper. SADDS including SAN would like to
become targeted drug delivery systems although only the phys-ical targeting to the MPS is achieved now. In the future, SAN
will be modified to got more functions such as long circulating,
To further evaluate the in vivo fate of acyclovir SAN, the tis-
sue distribution of SGSA was investigated. SGSA was mainlydistributed in liver, spleen and lung after acyclovir SAN bolusiv administration to rabbits SGSA content in liver was
4. Conclusions
very high to over 50% of the whole injected dose at 0.5 h, andmore than 30% at 4 h. SGSA concentration in liver, spleen and
SADDS are novel drug delivery approaches, and enlarge
lung was very high to more than 200 g/g at 0.5 h. Only a lit-
the fields of pharmaceutical researches. SADDS combine pro-
tle SGSA appeared in kidney (about 30 g/g at 0.5 h) and heart
drugs, molecular self-assembly with nanotechnology. Acyclovir
(about 4 g/g at 0.5 h). Nothing was found in brain. Therefore,
SAN in the paper become the successful examples of SADDS,
the nanosized SAN were mainly trapped by the MPS including
and show the well site-specific distribution in vivo. Much more
liver, spleen and lung. Also, lung capillary vessels could block
SADDS including SAN will be prepared from more hydrophilic
some large particles formed due to SAN aggregation. SGSA in
drugs such as nucleosides and lipids such glycerides on the basis
tissues was cleared in a first order kinetic mode and disappeared
of this paper. It can be predicted that SADDS will be useful to
completely at 48 h The elimination t1/2 of SGSA in
anti-viral, anti-cancer and gene therapy. Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207Acknowledgements
prodrug with activity against acyclovir-resistant herpes simplex virus. Proc. Natl. Acad. Sci. U.S.A. 90, 11835–11839.
This research is supported by the National Natural Science
Hostetler, K.Y., Beadle, J.R., Kini, G.D., Gardner, M.F., Wright, K.N., Wu,
T.H., Korba, B.A., 1997. Enhanced oral absorption and antiviral activity of
Foundation of China (30371700) and partly by Beijing Natural
1-O-octadecyl-sn-glycero-3-phospho-acyclovir and related compounds in
Science Foundation (7053074). Acknowledgements are given to
hepatitis B virus infection, in vitro. Biochem. Pharmacol. 53, 1815–1822.
Dr. Ying Tian and Dr. Jiannong Li for the discussion on antiviral
Ishida, T., Harashima, H., Kiwada, H., 2002. Liposome clearance. Biosci.
assay. We thank Dr. Gang-Jun Du of Henan University for his
Itojima, Y., Ogawa, Y., Tsuno, K., Hands, N., Yanagawa, H., 1992. Sponta-
neous formation of helical structures from phospholipid-nucleoside con-jugates. Biochemistry 31, 4757–4765. References
Jin, Y., Qiao, Y., Li, M., Ai, P., Hou, X., 2005. Langmuir monolayers of the
long-chain alkyl derivatives of a nucleoside analogue and the formation
Ahsan, F., Rivas, I.P., Khan, M.A., Suarez, A.I.T., 2002. Targeting to
of self-assembled nanoparticles. Colloid. Surf. B Biointerfaces 42, 45–51.
macrophages: role of physicochemical properties of particulate carriers—
Kim, I.S., Jeong, Y.I., Cho, C.S., Kim, S.H., 2000. Thermo-responsive self-
liposomes and microspheres—on the phagocytosis by macrophages. J.
assembled polymeric micelles for drug delivery in vitro. Int. J. Pharm.
Allen, T.M., Cullis, P.R., 2004. Drug delivery systems: entering the main-
Lambert, D.M., 2000. Rationale and applications of lipids as prodrug carriers.
Barenholz, Y., 2001. Liposome application: problems and prospects. Curr.
Langer, K., Balthasar, S., Vogel, V., Dinauer, N., von Briesen, H., Schu-
Opin. Colloid Interface Sci. 6, 66–77.
bert, D., 2003. Optimization of the preparation process for human serum
Bibby, D.C., Talmadge, J.E., Dalal, M.K., Kurz, S.G., Chytil, K.M., Barry,
albumin (HSA) nanoparticles. Int. J. Pharm. 257, 169–180.
S.E., Shand, D.G., Steiert, M., 2005. Pharmacokinetics and biodistribution
Lauraeus, S., Holopainen, J.M., Taskinen, M., Kinnunen, P.K.J., 1998. Aggre-
of RGD-targeted doxorubicin-loaded nanoparticles in tumor-bearing mice.
gation of dimyristoylphosphatidylglycerol liposomes by human plasma
low density lipoprotein. Biochim. Biophys. Acta 1373, 147–162.
Biron, K.K., Noblin, J.E., Miranda, P.D., Elion, G.B., 1982. Uptake, distri-
Li, J., Teng, L., Chen, H., Jiang, N., Jiang, J., 2003. Comparison of efficacies
bution, and anabolism of acyclovir in herpes simplex virus-infected mice.
of famciclovir with acyclovir against herpes simple virus type 1 and 2 in
Antimicrob. Agents Chemother. 21, 44–50.
vitro and in vivo. Chin. Pharm. J. 38, 423–426.
Choi, S.K., Vu, T.K., Jung, J.K., Kim, S.J., Jung, H.R., Chang, T., Kim,
Lipinski, C.A., Lombardo, F., Dominy, B.W., Feeney, P.J., 1997. Experimental
B.H., 2005. Nucleoside-based phospholipids and their liposomes formed
and computational approaches to estimate solubility and permeability in
drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25.
Davis, J.T., 2004. G-quartets 40 years later: from 5 -GMP to molecular
McGuigan, C., Slate, M.J., Parry, N.R., Perry, A., Harris, S., 2000. Synthesis
biology and supramolecular chemistry. Angew. Chem. Int. Ed. 43, 668–
and antiviral activity of acyclovir-5 -(phenyl methoxy alaninyl) phosphate
as a possible membrane-soluble nucleotide prodrug. Bioorg. Med. Chem.
Defrise-Quertain, F., Chatelain, P., Delmelle, M., Ruysschaert, J., 1984.
Model studies for drug entrapment and liposome stability. In: Grego-
Moghimia, S.M., Szebeni, J., 2003. Stealth liposomes and long circulat-
riadis, G. (Ed.), Liposome Technology, vol. 2. CRC Press, Florida, Boca
ing nanoparticles: critical issues in pharmacokinetics, opsonization and
protein-binding properties. Prog. Lipid Res. 42, 463–478.
de La Iglesia, P., Melo, S., Lopez, B., Rodriguez, M., Blanco, M.I., Mellado,
Moreau, L., Grinstaff, M.W., Barth´el´emy, P., 2005. Vesicle formation from a
P., de Ona, M., 1998. Rapid screening tests for determining in vitro
synthetic adenosine based lipid. Tetrahedron Lett. 46, 1593–1596.
susceptibility of herpes simplex virus clinical isolates. J. Clin. Microbiol.
Neyts, J., de Clercq, E., 1998. In vitro and in vivo inhibition of murine gamma
herpesvirus 68 replication by selected antiviral agents. Antimicrob. Agents
de Miranda, P., Blum, M.R., 1983. Pharmacokinetics of acyclovir after intra-
venous and oral administration. J. Antimicrob. Chemother. 12 (Suppl. B),
Ohki, S., Arnold, K., 2000. A mechanism for ion-induced lipid vesicle fusion.
Colloid. Surf. B Biointerfaces 18, 83–97.
Drummond, C.J., Fong, C., 2000. Surfactant self-assembly objects as novel
Opanasopit, P., Higuchi, Y., Kawakami, S., Yamashita, F., Nishikawa, M.,
drug delivery vehicles. Curr. Opin. Colloid Interface Sci. 4, 449–456.
Hashida, M., 2001. Involvement of serum mannan binding proteins and
Giorgi, T., Grepioni, F., Manet, I., Mariani, P., Masiero, S., Mezzina, E.,
mannose receptors in uptake of mannosylated liposomes by macrophages.
Pieraccini, S., Saturni, L., Spada, G.P., Gottarelli, G., 2002. Gel-like
Biochim. Biophys. Acta 1511, 134–145.
lyomesophases formed in organic solvents by self-assembled guanine rib-
Pastor-Anglada, M., Felipe, A., Casado, F.J., 1998. Transport and mode of
action of nucleoside derivatives used in chemical and antiviral therapies.
Glavas-Dodov, M., Fredro-Kumbaradzi, E., Goracinova, K., Simonoska, M.,
Trends Pharmacol. Sci. 19, 424–430.
Calis, S., Trajkovic-Jolevska, S., Hincal, A.A., 2005. The effects of
Prisbe, E.J., Martin, J.C., McGee, D.P.C., Barker, M.F., Smee, D.F., Duke,
lyophilization on the stability of liposomes containing 5-FU. Int. J. Pharm.
A.E., Matthews, T.R., Verheyden, J.P.H., 1986. Synthesis and antiher-
pes virus activity of phosphate and phosphonate derivatives of 9-[(1,3-
Gottarelli, G., Masiero, S., Mezzina, E., Pieraccini, S., Rabe, J.P., Samori, P.,
Dihydroxy-2-propoxy)methyl]guanine. J. Med. Chem. 29, 671–675.
Spada, G.P., 2000. The self-assembly of lipophilic guanosine derivatives
Qin, F.H., Xie, S.S., 1998. Establishment and characterization of murine bone
in solution and on solid surfaces. Chem. Eur. J. 6, 3242–3248.
marrow stromal macrophage line. Chinese Sci. Bull. 43, 178–186.
Groll, A.H., Mickiene, D., Werner, K., Petraitiene, R., Petraitis, V., Calen-
Ross, B.P., Braddy, A.C., McGeary, R.P., Blanchfield, J.T., Prokai, L., Toth,
dario, M., Field-Ridley, A., Crisp, J., Piscitelli, S.C., Walsh, T.J., 2000.
I., 2004. Micellar aggregation and membrane partitioning of bile salts,
Compartmental pharmacokinetics and tissue distribution of multilamel-
fatty acids, sodium dodecyl sulfate, and sugar-conjugated fatty acids: cor-
lar liposomal nystatin in rabbits. Antimicrob. Agents Chemother. 44,
relation with hemolytic potency and implications for drug delivery. Mol.
Harashima, H., Kiwada, H., 1996. Liposomal targeting and drug delivery:
Schreier, S., Malheiros, S.V.P., de Paula, E., 2000. Surface active drugs:
kinetic consideration. Adv. Drug Deliv. Rev. 19, 425–444.
self-association and interaction with membranes and surfactants. Physic-
Hostetler, K.Y., Parker, S., Sridhar, C.N., Martin, M.J., Li, J.L., Stuhmiller,
ochemical and biological aspects. Biochim. Biophys. Acta 1508, 210–234.
L.M., Wijk, G.M.T., Bosch, H., Gardner, M.F., Aldern, K.A., Richman,
Shah, J.C., Sadhale, Y., Chilukuri, D.M., 2001. Cubic phase gels as drug
D.D., 1993. Acyclovir diphosphate dimyristoylglycerol: a phospholipid
delivery systems. Adv. Drug Deliv. Rev. 47, 229–250. Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
Shameem, M., Imai, T., Otagiri, M., 1993. An in vitro and in vivo correlative
Zarif, L., 2002. Elongated supramolecular assemblies in drug delivery. J.
approach to the evaluation of ester prodrugs to improve oral delivery of
propranolol. J. Pharm. Pharmacol. 45, 246–252.
Zhai, L., Zhang, J., Shi, Q., Chen, W., Zhao, M., 2005. Transition from
Szoka, F., Papahadjopoulos, D., 1978. Procedure for preparation of liposomes
micelle to vesicle in aqueous mixtures of anionic/zwitterionic surfactants
with large internal aqueous space and high capture by reverse-phase evap-
studied by fluorescence, conductivity, and turbidity methods. J. Colloid
oration. Proc. Natl. Acad. Sci. U.S.A. 7, 4194–4198.
Torchilin, V.P., 2000. Drug targeting. Eur. J. Pharm. Sci. 11, S81–S91.
Zuidam, N.J., Lee, S.S., Crommelin, D.J., 1993. Sterilization of liposomes
Vaizoglu, M.O., Speiser, P.P., 1986. Pharmacosomes—a novel drug delivery
by heat treatment. Pharm. Res. 10, 1591–1596.
system. Acta Pharm. Suec. 23, 163–172.
Zuidam, N.J., Lee, S.S., Crommelin, D.J., 1995. Gamma-irradiation of
Whitesides, G.M., Grzybowski, B., 2002. Self-assembly at all scales. Science
non-frozen, frozen, and freeze-dried liposomes. Pharm. Res. 12, 1761–
PPTP y L2TP por Marisabel Rodriguez Bilardo Los protocolos PPTP y L2TP nacieron para crear VPNs. Además de permitir crear túneles a través de Internet, pueden encriptar los datos enviados y autenticar a los usuarios. Las VPNs conectan sitios remotos en forma segura y ademásbajan costos porque utilizan redes públicas en vez de líneasPPTP y L2TP son idénticos en la c
Women Realise Anaemia Can Be Treated Haemoglobin Levels Improve Through this intervention, more than a thousand women understood that theAfter six months of nutritious food and herbal treatment there was anweakness’ they experienced could be treated. They were able to examine theimprovement of up to two gram percentage in the haemoglobin level of 80 percentpallor of their eyes an


 International Journal of Pharmaceutics 309 (2006) 199–207
1. Properties and in vitro/in vivo behavior of acyclovir
Yiguang Jin , Li Tong , Ping Ai , Miao Li , Xinpu Hou
a Department of Pharmaceutical Chemistry, Beijing Institute of Radiation Medicine, Beijing 100850, PR China
b Department of Physical Pharmacy, School of Pharmaceutical Sciences, Peking University, Beijing 100083, PR China
c Department of Biochemistry and Biotechnology, College of Life Sciences, Beijing Normal University, Beijing 100875, PR China
Received 1 September 2005; received in revised form 8 November 2005; accepted 15 November 2005
Abstract
International Journal of Pharmaceutics 309 (2006) 199–207
1. Properties and in vitro/in vivo behavior of acyclovir
Yiguang Jin , Li Tong , Ping Ai , Miao Li , Xinpu Hou
a Department of Pharmaceutical Chemistry, Beijing Institute of Radiation Medicine, Beijing 100850, PR China
b Department of Physical Pharmacy, School of Pharmaceutical Sciences, Peking University, Beijing 100083, PR China
c Department of Biochemistry and Biotechnology, College of Life Sciences, Beijing Normal University, Beijing 100875, PR China
Received 1 September 2005; received in revised form 8 November 2005; accepted 15 November 2005
Abstract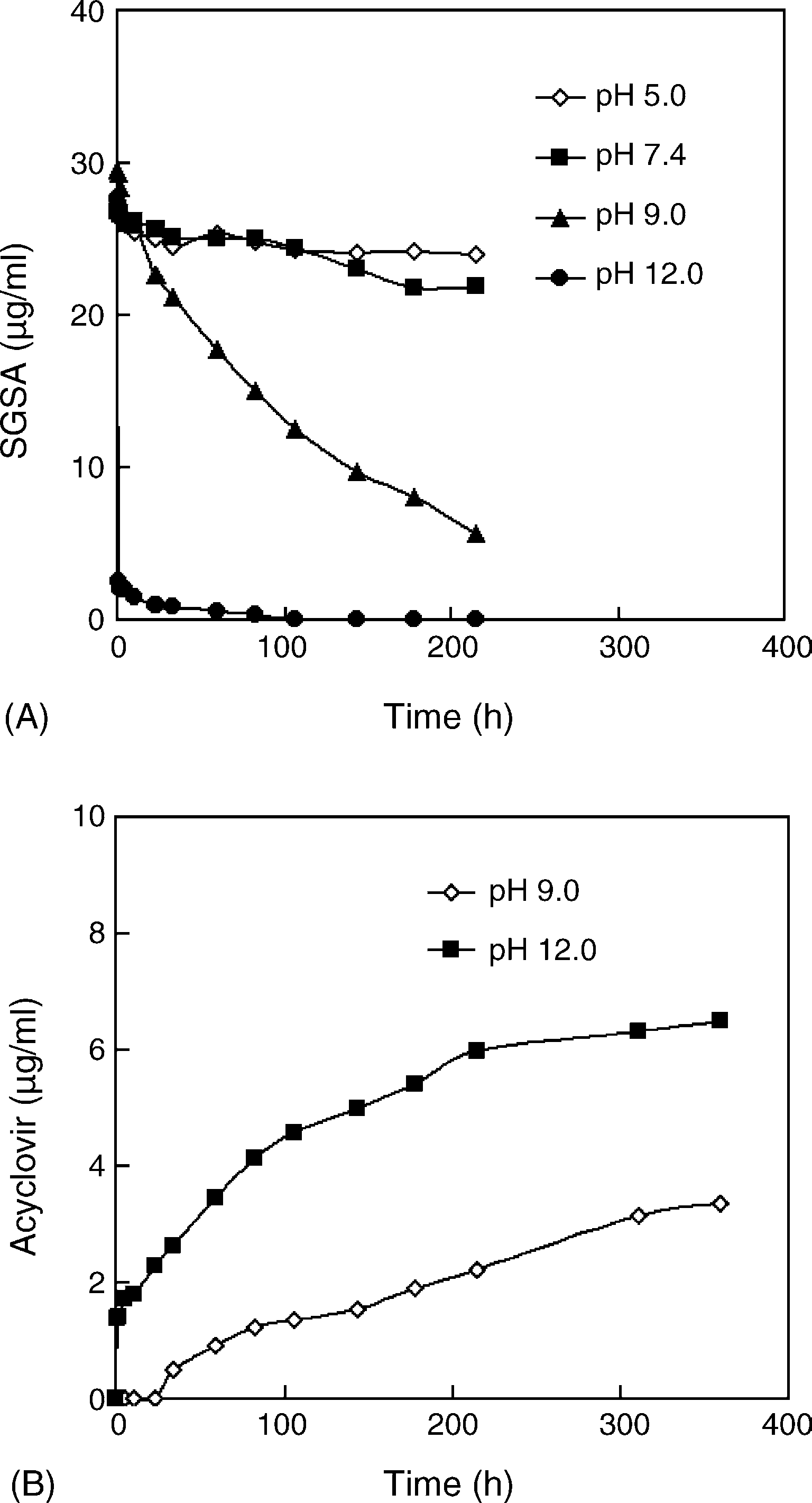
 Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
The autoclave or the 100 ◦C bath for sterilization neither influ-
Interestingly, after SGSA solutions in DMSO were diluted
enced the appearance of the SAN nor improved SGSA hydroly-
with buffers, SGSA degraded more rapidly than SGSA in SAN.
Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
The autoclave or the 100 ◦C bath for sterilization neither influ-
Interestingly, after SGSA solutions in DMSO were diluted
enced the appearance of the SAN nor improved SGSA hydroly-
with buffers, SGSA degraded more rapidly than SGSA in SAN.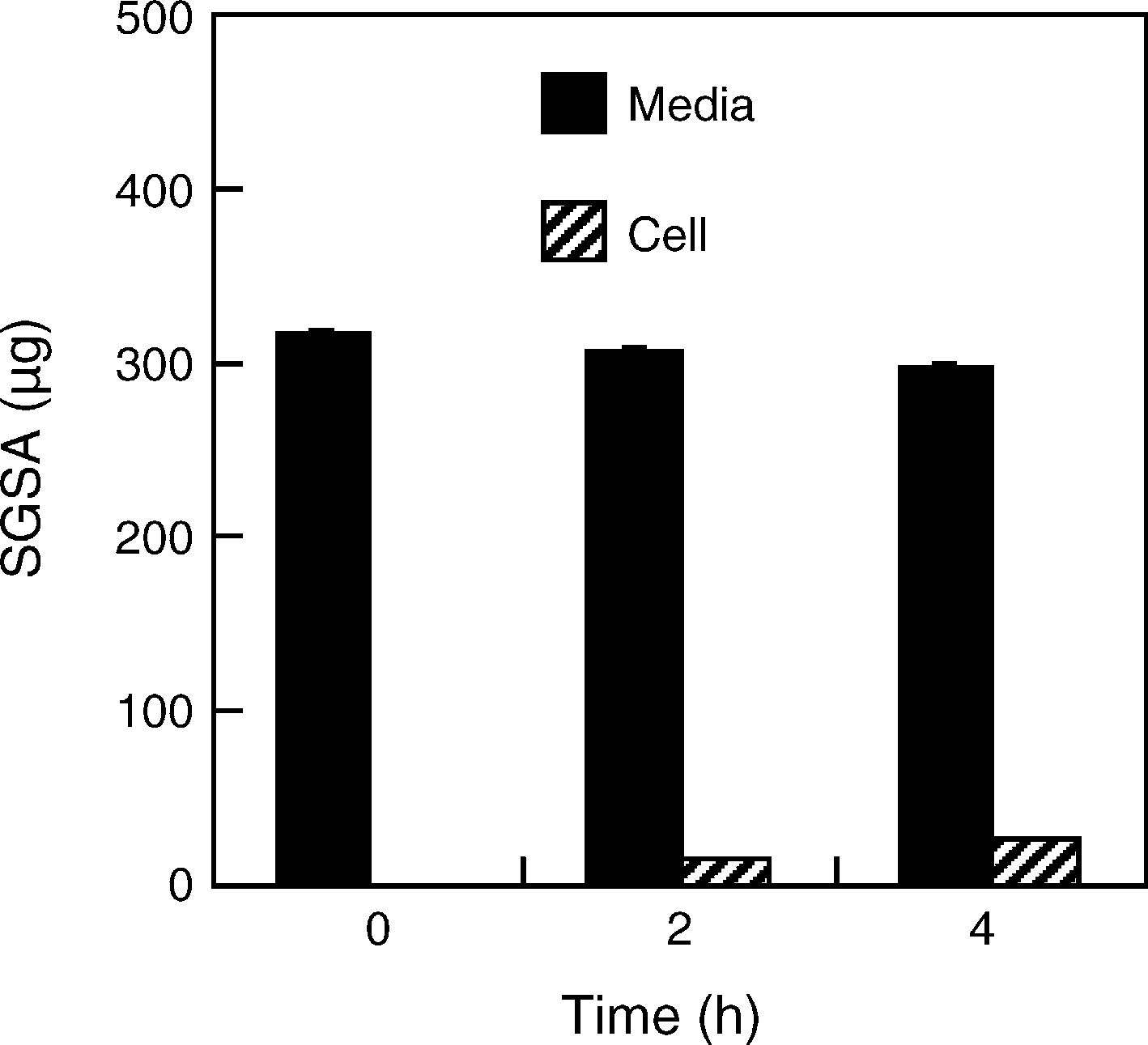 Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
of 25 h (Some intermediate SACV appeared in theplasma samples, which indicated that some enzymes in plasmacould hydrolyze the succinyl-glycerol ester bonds of SGSA. Thedegradation of SGSA in rabbit tissue homogenates depended onthe types of homogenates. The degradation t1/2 of SGSA in heart,liver, lung, spleen, kidney and brain were 5.0, 8.7, 11.8, 13.7,30.7 and 73.7 h, respectively, which must result from diverseenzyme activity between tissue homogenates.
Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
of 25 h (Some intermediate SACV appeared in theplasma samples, which indicated that some enzymes in plasmacould hydrolyze the succinyl-glycerol ester bonds of SGSA. Thedegradation of SGSA in rabbit tissue homogenates depended onthe types of homogenates. The degradation t1/2 of SGSA in heart,liver, lung, spleen, kidney and brain were 5.0, 8.7, 11.8, 13.7,30.7 and 73.7 h, respectively, which must result from diverseenzyme activity between tissue homogenates.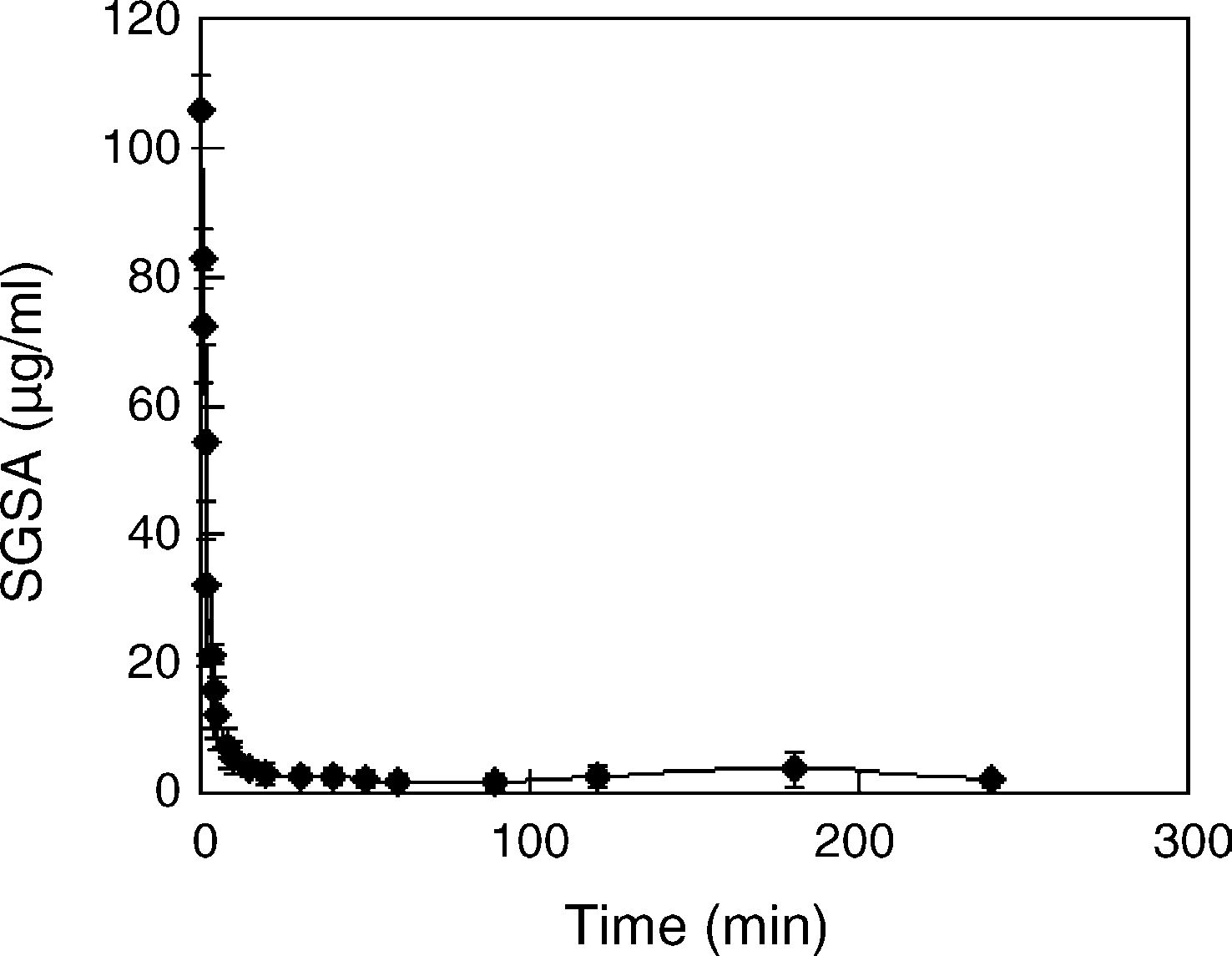
 Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
Fig. 4. The time profile of SGSA concentration in plasma after acyclovir SANbolus iv administration to rabbits with 30 mg SGSA/kg through ear vein. Results
Fig. 5. The tissue distribution of SGSA after acyclovir SAN bolus iv admin-
were expressed as the mean ± S.D. (n = 5). The field of 0–5 min was considered
istration to rabbits. Results were expressed as the mean ± S.D. (n = 3). SGSA
as the distribution phase with the half-life (t1/2␣) of 1.5 min, and 5–90 min as
was mainly distributed in liver, spleen and lung although only a little SGSA
the elimination phase with the half-life (t1/2) of 47 min based on the first order
appeared in kidney and heart, and nothing was found in brain. The elimination
kinetics. A little higher SGSA concentration at 180 min than the minimum level
t1/2 of SGSA in liver, spleen and lung were 7.6, 6.8 and 6.6 h, respectively, near to
was maybe due to some lung-blocked SAN-aggregating large particles releasing
the hydrolysis t1/2 of SGSA in tissue homogenates. The main tissue elimination
way of SGSA could be resulted from the metabolism.
Y. Jin et al. / International Journal of Pharmaceutics 309 (2006) 199–207
Fig. 4. The time profile of SGSA concentration in plasma after acyclovir SANbolus iv administration to rabbits with 30 mg SGSA/kg through ear vein. Results
Fig. 5. The tissue distribution of SGSA after acyclovir SAN bolus iv admin-
were expressed as the mean ± S.D. (n = 5). The field of 0–5 min was considered
istration to rabbits. Results were expressed as the mean ± S.D. (n = 3). SGSA
as the distribution phase with the half-life (t1/2␣) of 1.5 min, and 5–90 min as
was mainly distributed in liver, spleen and lung although only a little SGSA
the elimination phase with the half-life (t1/2) of 47 min based on the first order
appeared in kidney and heart, and nothing was found in brain. The elimination
kinetics. A little higher SGSA concentration at 180 min than the minimum level
t1/2 of SGSA in liver, spleen and lung were 7.6, 6.8 and 6.6 h, respectively, near to
was maybe due to some lung-blocked SAN-aggregating large particles releasing
the hydrolysis t1/2 of SGSA in tissue homogenates. The main tissue elimination
way of SGSA could be resulted from the metabolism.