Tadalafil gehört zur Gruppe der PDE5-Hemmer und wirkt über eine hochselektive Blockade des Enzyms Phosphodiesterase Typ 5. Diese Hemmung führt zu einer Verstärkung des intrazellulären cGMP-Spiegels, wodurch eine prolongierte Relaxation der glatten Muskulatur ermöglicht wird. Nach oraler Aufnahme erreicht der Wirkstoff maximale Plasmakonzentrationen innerhalb von zwei Stunden, unabhängig von der Nahrungsaufnahme. Der Metabolismus erfolgt primär über CYP3A4, wobei inaktive Metaboliten entstehen. Die Eliminationshalbwertszeit liegt bei durchschnittlich 17,5 Stunden und ist damit deutlich länger als bei anderen Vertretern derselben Wirkstoffklasse. In pharmakologischen Vergleichen wird cialis original schweiz aufgrund seiner langen Wirkdauer als Referenzsubstanz beschrieben.
Pt080000510p
0022-3565/00/2942-0510$03.00/0THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS
Copyright 2000 by The American Society for Pharmacology and Experimental Therapeutics
Hydroxyprolylserine Derivatives JBP923 and JBP485 Exhibitthe Antihepatitis Activities after Gastrointestinal Absorption inRats1
KE-XIN LIU, YUKIO KATO, TAI-ICHI KAKU, TOMOFUMI SANTA, KAZUHIRO IMAI, AKIRA YAGI, TAKASHI ISHIZU, andYUICHI SUGIYAMA
Graduate School of Pharmaceutical Sciences, University of Tokyo, Hongo, Bunkyo-ku, Tokyo (K.L., Y.K., T.S., K.I., Y.S.); Japan BioproductsIndustry Co. Ltd., Tomigaya, Shibuya-ku, Tokyo (T.K.); and Faculty of Pharmacy and Pharmaceutical Sciences, Fukuyama University,Gakuencho, 1, Fukuyama, Hiroshima (A.Y., T.I.), Japan
This paper is available online at http://www.jpet.org
ABSTRACT It has been a desire to develop orally effective therapeutic
effect on hepatocytes because glutamic-oxaloacetic transam-
agents that restore the liver function in chronic injury. Here we
inase and lactate dehydrogenase activities in the medium of
demonstrated that trans-4-L-hydroxyprolyl-L-serine (JBP923)
hepatotoxin-exposed primary cultured hepatocytes were re-
and cyclo-trans-4-L-hydroxyprolyl-L-serine (JBP485), which
duced by these compounds. When comparing the plasma con-
was previously isolated from hydrolysate of human placenta,
centration-time profile of JBP923 after its i.v., oral, and portal
exhibit potent antihepatitis activity after their oral administra-
vein injection, it is suggested that JBP923 is almost completely
tion. The increase in bilirubin concentration and activities of
absorbed from gastrointestinal lumen, and hepatic first-pass
liver cytosolic enzymes in serum caused by ␣-naphthylisothio-
removal is minor. JBP923 inhibited the proton-dependent
cyanate intoxication in rats were significantly countered both
transport of glycylsarcosine in brush-border membrane vesi-
after i.v. and oral administration of these dipeptides, whereas
cles, suggesting that peptide transport system(s) may recog-
glycyrrhizin, which has been used in the treatment of chronic
nize JBP923. Thus, these dipeptides are potent antihepatitis
hepatitis, is active only after its i.v. administration. Antihepatitis
reagents that are still active after oral administration and may
activity of dipeptides results, at least partially, from their direct
be useful for clinical applications.
Several types of drugs have been used to treat chronic
was first isolated from Laennec, a trade name for the hydro-
hepatitis and cirrhosis. These include prednisone and aza-
lysate of human placenta, as mitogens for a baby hamster
thioprine for the treatment of autoimmune chronic hepatitis
kidney cell line, and subsequently it has been enantioselec-
(Bellary et al., 1995; Czaja, 1999) and interferons for viral
tively synthesized by chemical means (Yagi et al., 1998).
hepatitis (Dumoulin et al., 1999; Par et al., 1999; Shiffman et
Laennec is produced by Japan Bioproducts Industry Co. Ltd.
al., 1999). To improve the liver function in chronic hepatitis,
(Tokyo, Japan) by purification of human placental extracts
glycyrrhizin, one of the main constituents of Glycyrrhiza
involving dialysis, heat treatment, and hydrolysis. Laennec
glabra L, which has antiallergic, anti-inflammatory, and an-
has been clinically used to treat chronic hepatic injuries for
tihepatitis activities, is frequently used (Nose et al., 1994,
over forty years in Japan. Recently we found that Laennec
1996; Wang et al., 1994; Takeda et al., 1996; Arase et al.,
stimulates liver regeneration and decreases cytosolic enzyme
1997). However, glycyrrhizin is usually administered i.v. be-
[glutamic-pyruvic transaminase (GPT), alkaline phospha-
cause it is inactive after oral administration. Therefore, for
tase (ALP), leucine aminopeptidase (LAP), ␥-glutamyltrans-
the treatment of chronic liver injuries, orally effective ther-
ferase (␥-GTP)] activities in serum in ␣-naphthylisothiocya-
apeutic agents have to be developed.
nate (ANIT)-intoxicated rats (Liu et al., 1995). We have
cyclo-trans-4-L-Hydroxyprolyl-L-serine (JBP485) (Fig. 1)
found that Laennec also contains trans-4-L-hydroxyprolyl-L-serine (JBP923). Those findings prompted us to synthesize
Received for publication February 8, 2000. 1
these dipeptides and investigate their protecting effect on
This study was supported in part by a Grant-in-Aid for Scientific Research
provided by the Ministry of Education, Science and Culture of Japan.
hepatocytes from hepatotoxin treatment. ABBREVIATIONS: JBP485, cyclo-trans-4-L-hydroxyprolyl-L-serine; JBP923, trans-4-L-hydroxyprolyl-L-serine; ANIT, ␣-naphthylisothiocyanate; TFA, trifluoroacetic acid; BIL, bilirubin concentration; GOT, glutamic-oxaloacetic transaminase; GPT, glutamic-pyruvic transaminase; LAP, leucine aminopeptidase; LDH, lactate dehydrogenase; ALP, alkaline phosphatase; DMSO, dimethyl sulfoxide; BBMVs, brush-border membrane vesicles; Gly-Sar, glycylsarcosine; ␥-GTP, ␥-glutamyltransferase. Dipeptides Orally Exhibiting Antihepatitis Activities
as described previously (Kato et al., 1994). Briefly, isolated hepato-cytes suspended in Williams’ medium E supplemented with 5% calfserum, 10Ϫ9 M insulin, and 10Ϫ9 M dexamethasone were plated onto24-well plastic dishes coated with type I collagen. The nonattachedcells were removed by washing with fresh culture medium at 3 hafter plating. CCl , first dissolved in dimethyl sulfoxide (DMSO) at
1.0 M, was diluted with fresh medium containing JBP923, JBP485,glycyrrhizin, or 18-glycyrrhetinic acid (Sigma) to give a final con-centration of 5 mM. Control experiments were performed in thepresence of only DMSO. At 24 h after plating, the medium wasreplaced with the buffer containing both CCl and an appropriate
drug. The monolayers were further cultured for 24 h, and culturemedium was collected and centrifuged at 24,000g for 20 min. GOTand lactate dehydrogenase (LDH) were assayed as described above. One unit was defined as the amount of activity catalyzing formationof 1 mol of product/1 min. Pharmacokinetic Analysis in Normal Rats. Under ether an- Fig. 1. Chemical structures of JBP923 and JBP485.
esthesia, JBP923 (3.13 or 25 mg/kg) dissolved in saline was admin-
Although both JBP923 and JBP485 have simple chemical
istered through the penis vein, through the portal vein, or into the
structures with dipeptide backbone (Fig. 1), here we report
stomach with a gastric sonde. This ether anesthesia was sufficient to
their potent antihepatotoxic activity in rats. It is notable that
allow portal vein injection, which was performed over a period of 10
these compounds are active in vivo after oral administration.
min using an infusion pump. Plasma was collected from the externaljugular vein at the indicated times, and the JBP923 concentration in
To support our hypothesis that these dipeptides are orally
plasma was determined by HPLC as described below. The plasma
absorbed and directly interact with hepatocytes to restore
concentration (Cp)-time profiles of JBP923 after i.v. and oral admin-
their functions, we investigated the gastrointestinal absorp-
istration were fitted to the following equations, respectively:
tion in vivo and antihepatitis activity in primary culturedhepatocytes. Our findings demonstrate that these dipeptides
Cp ϭ A exp(Ϫ␣t) ϩ B exp(Ϫt)
may be applicable as oral drugs for the treatment of liver
aFexp(Ϫkat)͑A͑1 Ϫ exp(Ϫ͑␣ Ϫ ka
Experimental Procedures
where k and F were absorption rate constant and bioavailability,
Animals and Materials. Male Wistar rats weighing 250 and
150 g (Nisseizai, Tokyo, Japan) for in vivo and in vitro studies,
respectively, were used throughout the experiments. All animals
were treated humanely. The studies reported in this article have
is area under the plasma concentration-time profile
been carried out in accordance with the Guide for the Care and Useof Laboratory Animals as adopted and promulgated by the NationalInstitutes of Health. JBP923 and JBP485 were synthesized by Wa-
tanabe Chemical Industries Company (Hiroshima, Japan). Acetoni-
The hepatic availability (F ) was calculated as:
trile, tetrahydrofuran, trifluoroacetic acid (TFA), dioxane, and dis-
tilled water, all of HPLC grade, were purchased from Wako Pure
Chemical Industries (Osaka, Japan). 4-Fluoro-7-nitro-2,1,3-benzox-adiazole was from Tokyo Kasei Co. (Tokyo, Japan).
was AUC after portal vein injection. The AUC during
Antihepatotoxic Activities In Vivo. ANIT (Sigma, St. Louis,
the 10-min portal vein infusion was calculated by trapezoidal rule.
MO) dissolved in olive oil was injected i.p. at a dose of 50 mg/kg body
The AUC after the end of infusion was obtained by eq. 4, where the
wt. For i.v. administration, JBP923 and JBP485 (25 mg/ml), dis-
plasma concentration-time profile was fitted also to eq. 1. The input
solved in saline, or glycyrrhizin injection (Minophagen Pharmaceu-
data for all the fitting were weighted as the reciprocal of the square
tical Co., Tokyo, Japan) were administered through the penis vein.
of the observed values, and the algorithm used for the fitting was the
For oral administration, JBP923 and JBP485 dissolved in saline or
glycyrrhizin tablets (Minophagen Pharmaceutical Co.) dissolved in
Determination of JBP923 in Plasma by HPLC. To 12.5 l of
5% glucose solution were administered via esophagus with a gastric
plasma, 500 l of methanol was added, and the mixture was centri-
sonde. Administrations of these drugs were performed at 30 min
fuged at 600g for 5 min for deproteinization. The supernatant was
before and 8, 22, 32, and 46 h after ANIT treatment. All the admin-
collected and dried under reduced pressure using a centrifugal evap-
istrations were performed under ether anesthesia. Serum was col-
orator. For the derivatization of an imino group in JBP923, 20 l of
lected 48 h after ANIT treatment. For serum collection, rats were
50 mM borate buffer (pH ϭ 8.0) and 30 l of 20 mM 4-fluoro-7-nitro-
anesthetized with ether, and approximately 10 ml of blood was
2,1,3-benzoxadiazole dissolved in acetonitrile were added to the
sampled from the aorta abdominalis. Blood was then left on ice for 20
dried sample (Fukushima et al., 1995). The reaction mixture was
min and centrifuged at 1000g for 5 min to obtain the supernatant as
heated at 60°C for 5 min, and 450 l of 1% TFA in water was added
serum. The total bilirubin concentration (BIL) and activity of liver-
to the mixture to stop the reaction. Twenty microliters of the result-
specific cytosolic enzymes, such as GPT, LAP, ALP, glutamic-oxaloa-
ant solution was subjected to HPLC analysis. The HPLC system
cetic transaminase (GOT) and ␥-GTP, in the rat serum were deter-
consisted of a model L-6320 Intelligent pump (Hitachi, Tokyo, Japan)
mined using the appropriate assay kits (Wako Pure Chemical
and a model F-1050 fluorescence spectrophotometer (Hitachi). An
ODS-COSMOSIL (4.6 ϫ 150 mm, i.d.) column (Nacalai Tesque, To-
Determination of Biochemical Marker Leakage from Pri-
kyo, Japan) was used. The excitation and emission wavelengths were
mary Cultured Rat Hepatocytes. Parenchymal hepatocytes were
fixed at 470 and 540 nm, respectively. A gradient HPLC system was
plated at a density of 1.25 ϫ 105 cells/1.88 cm2 and cultured for 24 h
adopted. Eluent A, water/acetonitrile (95.5:4.5, v/v) containing 4.5%
Liu et al.
dioxane, 1% tetrahydrofuran, and 0.05% TFA, and eluent B (aceto-
represent the transport velocity in the presence
nitrile) were used. The elution program was as follows: eluent A, 100
and absence of inhibitor, respectively; I is the inhibitor concentra-
to 0% from 0 to 18 min; eluent B, 100 to 0% from 18.1 to 35 min;
tion. Equation 6 is based on the assumption of competitive inhibition
eluent A, 100% from 35.1 to 45 min. The flow rate was 1 ml/min.
in a case when the Michaelis constant (K ) is much higher than the
Brush-Border Membrane Vesicles
substrate concentration. In a preliminary study, we found that the
(BBMVs). BBMVs were prepared by the method of Kessler (Kessler K for Gly-Sar uptake was 15.5 mM in rabbit BBMVs, and therefore
et al., 1978). Briefly, the approximately 50-cm proximal portion of
the substrate concentration chosen for this experiment was 0.668
the jejunum was isolated from male rabbits (2.0 –2.5 kg; Nisseizai).
The mucosa was scraped off and homogenized in a volume of ice-cold
Statistical Analysis. Statistical analysis was performed by Stu-
buffer A (2 mM Tris/HEPES buffer containing 50 mM D-mannitol,
dent’s t test to identify significant differences between various treat-
pH ϭ 7.1). The homogenization was carried out with a Waring
blender for 2 min at a speed of 18,000 rpm. Solid CaCl was added to
the homogenate to give a final concentration of 10 mM, and the
mixture was stirred in an ice bath for 15 min. It was then centrifugedat 500g for 15 min, and the supernatant was centrifuged at 1500g for
Antihepatotoxic Effect of JBP923 and JBP485 in
30 min. The pellet was homogenized with buffer A in a glass/Teflon
ANIT-Intoxicated Rats. To examine whether JBP923 and
Potter homogenizer at a speed of 1000 rpm. The mixture then was
JBP485 promote the repair of injured liver function in ANIT-
centrifuged at 750g for 30 min. The pellet was homogenized in a
intoxicated rats, we determined the change in BIL and ac-
glass/Teflon Potter homogenizer again with the same buffer and the
tivities of liver cytosolic enzymes in serum of ANIT-intoxi-
speed mentioned above. The supernatant was centrifuged at 48,000gfor 30 min, and then the pellet was suspended with a 23-gauge
cated rats after administration of JBP923 and JBP485 (Table
needle. The centrifugation was performed again with the same speed
1). The increase in BIL and liver cytosolic enzyme activities
and time, and then the pellet was suspended with a 27-gauge needle.
caused by ANIT intoxification were countered by i.v. and oral
Finally, protein concentration in the suspension was determined
administration of JBP923 and JBP485 (Table 1). The reduc-
using a Bio-Rad protein assay kit with BSA as a standard; the
tion in all the marker values were significant at i.v. and oral
concentration was 25 mg/ml with transport buffer (10 mM Tris/
doses of more than 1.36 and 25 mg/kg, respectively, both for
HEPES buffer with 270 mM D-mannitol, pH ϭ 7.5).
JBP923 and JBP485 (Table 1). When the i.v. and oral doses
Uptake of [14C]glycylsarcosine (Gly-Sar) by BBMVs was measured
were increased up to 6.25 and 25 mg/kg, respectively, the
by the rapid filtration method described by Hopfer (Hopfer et al.,
bilirubin, LAP, and ALP levels were almost comparable with
1973). The uptake was started by adding 4 l of BBMVs (100 g) to
those values in normal rats (Table 1).
16 l of transport buffer (20 mM Tris-citrate buffer, pH ϭ 5.5)containing JBP923, JBP485, and [14C]Gly-Sar at 37°C. The final
Comparison of the Antihepatotoxic Effect of JBP923
substrate concentration was 68 and 600 M [14C]Gly-Sar (2.96 GBq/
and JBP485 with Glycyrrhizin. Because glycyrrhizin is
mmol) and unlabeled Gly-Sar, respectively. The reaction was
one of the most frequently administered drugs in chronic
stopped at the desired time by adding 1 ml of ice-cold stop buffer
liver-injured patients, the antihepatitis activity was com-
(pH ϭ 7.5), which contained 20 mM Tris, 20 mM HEPES, and 300
pared among JBP923, JBP485, and glycyrrhizin (Fig. 2).
mM mannitol. Then, 0.9 ml of the diluted sample was applied im-
Intravenous or oral administration of JBP923 caused the
mediately on a Millipore filter (HAWP, 0.45-m pore size) and
reduction of GPT at almost the same doses (Fig. 2A). The
washed rapidly twice with 5 ml of ice-cold stop buffer. The uptake of
reduction in GPT activity was also observed after i.v. or oral
[14C]Gly-Sar by BBMVs trapped on the Millipore filter was mea-
administration of JBP485, although oral administration ex-
sured in a liquid scintillation spectrometer. The inhibition constant
hibited weaker antihepatitis activity (Fig. 2B). Minimal re-
(K ) was obtained by fitting the data to the following equation:
duction was found after oral administration of glycyrrhizin,
whereas its i.v. administration decreased GPT level (Fig. 2C).
TABLE 1Change in BIL and activity of liver cytosolic enzymes in serum in ANIT-intoxicated rats treated with JBP923 and JBP485Each value represents the mean Ϯ S.E. of three animals.
* P Ͻ .05, ** P Ͻ .01, significantly different from ANIT-treated rats. Dipeptides Orally Exhibiting Antihepatitis Activities Fig. 2. Comparison of antihepatitis effect on ANIT-intoxicated rats among JBP923 (A), JBP485 (B), and glycyrrhizin (C). JBP923, JBP485, or glycyrrhizin was administered through the penis vein (F) or orally (E) 30 min before and 8, 22, 32, and 46 h after ANIT treatment. Serum was collected at 48 h, and the activity of GPT was determined. The values are expressed as means Ϯ S.E. of three rats. *P Ͻ .05; **P Ͻ .01, significantly different from saline alone.
Thus, these dipeptides exhibit antihepatotoxic effect after
pass elimination of JBP923, its plasma concentration-time
oral administration, but glycyrrhizin does not.
profiles in rats were determined after i.v., oral, and portal
Antihepatotoxic Effect on Primary Cultured Hepa-
vein administrations (Fig. 4). The plasma concentration of
tocytes. The decrease in leakage of liver cytosolic enzyme by
JBP923 was gradually decreased after i.v. administration
these compounds was also examined in vitro in primary
with a terminal phase half-life of 21 to 24 min (Fig. 4). The
cultured hepatocytes intoxicated with CCl (Fig. 3). The GOT
gastrointestinal absorption of JBP923 was rapid with a k of
activity in the medium was decreased by addition of JBP923
0.01 to 0.04 minϪ1 and maximum plasma concentration ob-
and JBP485 in a concentration-dependent manner, the GOT
served within 30 min (Fig. 4 and Table 2). The AUC after oral
activity at the highest concentration being almost compara-
administration was almost comparable with that after i.v.
ble with that of hepatocytes without CCl intoxication (Fig.
administration both at 3.13 and 25 mg/kg (Table 2), suggest-
3A). Both glycyrrhizin and 18-glycyrrhetinic acid also de-
ing almost complete oral absorption. The AUC after portal
creased the GOT activity, although such effect at concentra-
vein administration was also comparable with that after i.v.
tions greater than 50 M was smaller than JBP923 and
administration at 3.13 mg/kg (Table 2), suggesting that he-
JBP485 (Fig. 3A). The reduction in LDH activity was found
patic first-pass elimination is not so remarkable.
in the presence of any compounds examined (Fig. 3B). Such
Effect of JBP923 and JBP485 on Uptake of [14C]Gly-
reduction in the presence of JBP923 or JBP485 was found at
Sar in Intestinal BBMVs. To examine the interaction of
a lower concentration than that found in the presence of
these dipeptides with oligopeptide-specific transporters ex-
glycyrrhizin or 18-glycyrrhetinic acid.
pressed in small intestines, their inhibitory effect on uptake
Pharmacokinetics of JBP923 in Normal Rats. To ex-
of Gly-Sar, a typical substrate of peptide transporter PEPT1,
amine the gastrointestinal absorption as well as hepatic first-
by rabbit intestinal BBMVs was investigated (Fig. 5). The
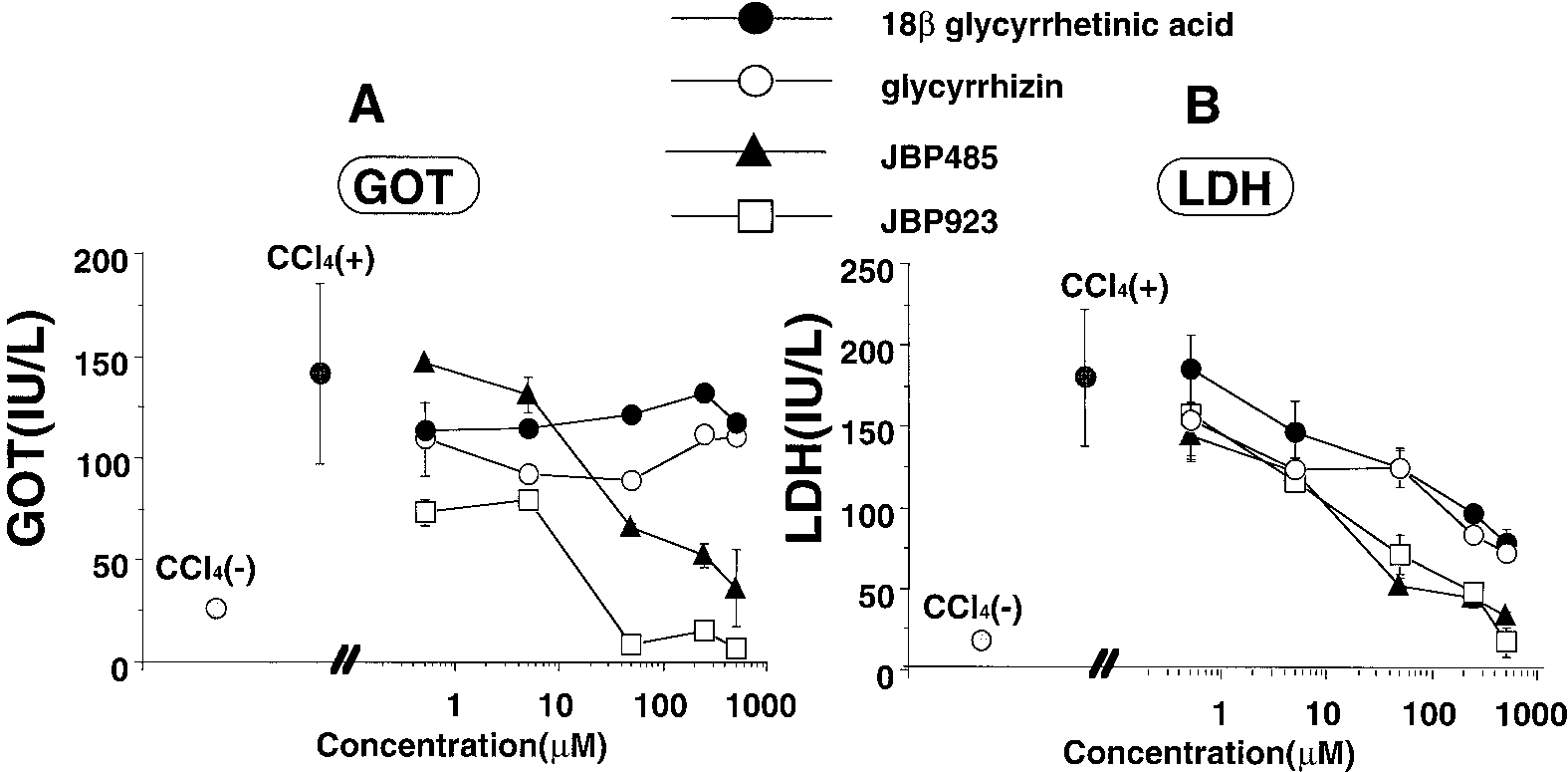
Fig. 3. The recovery of the GOT and LDH leakage by JBP923, JBP485, glycyrrhizin, or 18-glycyrrhetinic acid in the medium of primary cultured rat hepatocytes exposed to CCl . Parenchymal hepatocytes were cultured for 24 h, then the cultured medium was changed to the fresh medium containing
5 mM CCl (control) or 5 mM CCl with various concentrations of the indicated compounds. Hepatocytes were further cultured for 24 h, and the
activities of GOT and LDH in the medium were determined. CCl (Ϫ), medium without treatment of CCl ; CCl (ϩ), medium with CCl in 0.45% of
DMSO; DMSO, medium containing DMSO alone. The values are expressed as means Ϯ S.E. of three rats. Liu et al. Fig. 4. Plasma concentration-time profiles of JBP923 after i.v., portal vein, and oral administrations in rats. JBP923 at 3.13 (A) or 25 (B) mg/kg was administered through the penis vein (F), portal vein (Œ), and orally (E) to rats. Plasma was collected from the external jugular vein at the indicated time, and the JBP923 concentration in plasma was determined by HPLC. Points are expressed as means Ϯ S.E. of three to five rats.
TABLE 2Pharmacokinetic parameters of JBP923 in rats
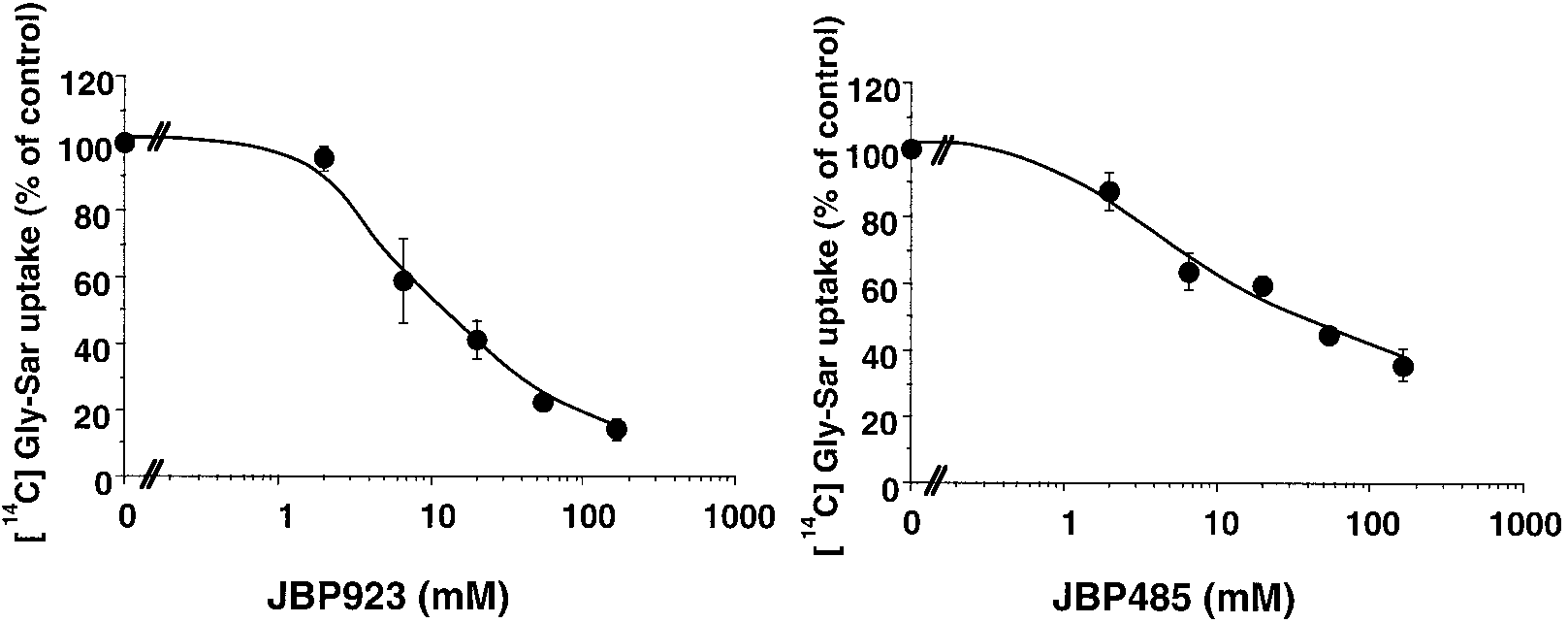
pv, portal vein. a Bioavailability (F) obtained from eq. 2. b Hepatic availability (Fh) obtained from eq. 5. c Significantly different (P Ͻ .05). Fig. 5. Inhibitory effects of JBP923 and JBP485 on [14C]Gly-Sar uptake by rabbit intestine BBMVs. Membrane vesicles were preloaded with transport buffer (pH ϭ 7.5). Uptake of [14C]Gly-Sar (68 M) with cold Gly-Sar (600 M) in 20 mM Tris-citrate transport buffer (pH ϭ 5.5) was measured at 37°C for 2 min as a control. After incubating the membrane vesicles containing various concentrations of JBP923 and JBP485 at 37°C for 2 min, the uptake of [14C]Gly-Sar (68 M) with cold Gly-Sar (600 M) was measured. Points are expressed as means Ϯ S.E. of three independent experiments.
uptake of [14C]Gly-Sar exhibited proton dependence because
The antihepatitis activity of JBP923 was observed both after
the uptake was higher in medium at pH 5.5 than that in
its i.v. and oral administration. This was compatible with our
medium at pH 7.5 (data not shown). Both JBP923 and
finding that gastrointestinal absorption of JBP923 is almost
JBP485 inhibited the uptake of [14C]Gly-Sar in a concentra-
complete (Table 2). JBP923 decreased both GOT and LDH
tion-dependent manner (Fig. 5) with K values of 13 and 31
activity in the medium of in vitro primary cultured hepato-
mM for JBP923 and JBP485, respectively.
cytes (Fig. 3). Such direct effect on hepatocytes was found atthe concentrations above ϳ50 M. On the other hand, the
Discussion
plasma JBP923 concentration after its oral administration at
In this study, we found two dipeptide compounds, JBP923
25 mg/kg was higher than 50 M (10 g/ml) until at least 2 h
and JBP485, that decrease BIL and hepatic cytosolic en-
(Fig. 4B). This means that effective JBP923 concentration
zymes activities in serum of ANIT-intoxicated rats (Table 1).
was maintained after oral administration. Thus, it seems to
Dipeptides Orally Exhibiting Antihepatitis Activities
be that JBP923 showed pharmacological activity after its
these activities are actually related to its protective effect on
gastrointestinal absorption and subsequent interaction with
liver function (Yi et al., 1996; Liu et al., 1998; Shaikh et al.,
hepatocytes, although this finding does not deny the possi-
1999). Therefore, the mechanism of antihepatitis activity of
bility of the existence of its active metabolites.
these two dipeptides should also be clarified to further dem-
Although glycyrrhizin also decreased the liver function
onstrate their applicability to clinical stages.
marker enzyme both in vivo and in vitro (Figs. 2 and 3), itspharmacological effect was minimal after oral administra-
References
tion (Fig. 3C). This is compatible with the fact that glycyr-
Arase Y, Ikeda K, Murashima N, Chayama K, Tsubota A and Koida I (1997) The long
rhizin was usually administered i.v. for the treatment of
term efficacy of glycyrrhizin in chronic hepatitis C patients. Cancer 79:1494 –1500.
chronic hepatic injuries. Glycyrrhizin was reported to be
Bellary S, Schiano T, Hartman G and Black M (1995) Chronic hepatitis with
combined features of autoimmune chronic hepatitis and chronic hepatitis C: Fa-
hydrolyzed by bacteria in the stomach and large intestinal
vorable response to prednisone and azathioprine. Ann Int Med 123:32–34.
content, and the first-pass elimination might be the reason
Czaja AJ (1999) Drug therapy in the management of type 1 autoimmune hepatitis. Drugs 57:49 – 68.
for its low bioavailability (Wang et al., 1994; Takeda et al.,
Dumoulin FL, Leifeld L, Sauerbruch T and Spengler U (1999) Autoimmunity in-
1996). Wang et al. (1994) reported that glycyrrhizin was
duced by interferon-alpha therapy for chronic viral hepatitis. Biomed Pharmaco- ther 53:242–254.
metabolized to 18-glycyrrhetinic acid. Nose et al. (1994)
Fukushima T, Kato M, Santa T and Imai K (1995) Enantiomeric separation and
suggested that 18-glycyrrhetinic acid was more potent than
sensitive determination of D, L-amino acids derivatized with fluorogenic benzo-
glycyrrhizin in terms of its antihepatotoxic activity toward
furazan reagents on Pirkle type stationary phases. Biomed Chromatogr 9:10 –17.
Gannapathy Y, Burckhardt G and Leibach FH (1984) Characteristics of glycylsar-
CCl -treated primary cultured hepatocytes. Also in this
cosine transport in rabbit intestinal brush-border membrane vesicles. J Biol Chem
study, 18-glycyrrhetinic acid decreased LDH activity in the
259:8954 – 8959.
Guo A, Hu P, Balimane PV, Leibach FH and Sinko PJ (1999) Interactions of a
medium of cultured hepatocytes (Fig. 3). This active metab-
nonpeptidic drug, valacyclovir, with the human intestinal peptide transporter
olite was also found in plasma of humans after oral admin-
(hPEPT1) expressed in a mammalian cell line. J Pharmacol Exp Ther 289:448 – 454.
istration of 100 mg of glycyrrhizin, although its concentration
Hopfer U, Nelson K, Perrotto J and Isselbacher KJ (1973) Glucose transport in
was at most 0.5 g/ml, corresponding to 1.1 M, which was
isolated brush border membrane from rat small intestine. J Biol Chem 248:25–32.
Kato Y, Liu KX, Nakamura T and Sugiyama Y (1994) Heparin-hepatocyte growth
less than the effective concentration found in this study and
factor (HGF) complex with low plasma clearance and retained hepatocyte prolif-
the report by Nose et al. (1996). Thus, oral administration of
erating activity. Hepatology 20:417– 422.
Kessler M, Acuto O, Storelli C, Murer H, Muller M and Semenza G (1978) A modified
glycyrrhizin does not exhibit clear antihepatotoxic activity,
procedure for the rapid preparation of efficiently transporting vesicles from small
and it is anticipated that orally active antihepatitis drugs
intestinal brush border membrane. Biochim Biophys Acta 506:136 –154.
Kitagawa S, Takeda J and Sato S (1999) pH-dependent inhibitory effects of angio-
will be developed for the clinical treatment of chronic hepa-
tensin-converting enzyme inhibitors on cefroxadine uptake by rabbit small intes-
tinal brush-border membrane vesicles and their relationship with hydrophobicity and the ratio of zwitterionic species. Biol Pharm Bull 22:721–724.
JBP923 exerts antihepatitis activity after oral administra-
Li J and Hidalgo IJ (1996) Molecular modeling study of structural requirements for
tion (Table 1). JBP923 was found in plasma only 1 min after
the oligopeptide transporter. J Drug Target 4:9 –17.
its oral administration (Fig. 4). This finding as well as its
Liu KX, Okazawa I and Kaku T (1995) Effect of Laennec on liver regeneration (in
Japanese). Clin Pharmacol Ther 5:2187–2194.
complete gastrointestinal absorption suggests that a certain
Liu W, Kato M, Akhand AA, Hayakawa A, Takemura M,. Yoshida S, Suzuki H and
specific mechanism may contribute to the gastrointestinal
Nakashima I (1998) The herbal medicine sho-saiko-to inhibits the growth ofmalignant melanoma cells by upregulating Fas-mediated apoptosis and arresting
transport. It also should be noted that the k was signifi-
cell cycle through downregulation if cyclin dependent kinases. Int J Oncol 12:
cantly lower at 25 mg/kg than at 3.13 mg/kg, suggesting the
Nose M, Ito M, Kaminura K, Shimizu M and Ogihara Y (1994) A comparison of the
slower absorption at a higher dose. It has been reported that
antihepatotoxic activity between glycyrrhizin and glycyrrhetinic acid. Planta Med
certain hydrophilic -lactam antibiotics can be transported
60:136 –139.
Nose M, Ito M, Kiode T, Terawaki K and Ogihara Y (1996) The effect of 18a,
by oligopeptide transporters in gastrointestinal tissues. This
-glycyrrhetinic acid on the hormonal induction of tyrosine aminotransferase in
transport system(s) also accepts other types of pharmaceuti-
rat primary cultured hepatocytes. Planta Med 62:410 – 413.
Par A, Telegdy L, Gogl A and Muller E (1999) Interferon therapy of chronic viral
cal agents such as angiotensin-converting enzyme inhibitors,
hepatitis in Hungary: 5-year experience. A multicenter study. Orv Hetil 140:1227–
renin inhibitors, and thrombin inhibitors (Gannapathy et al.,
Shaikh ZA, Vu TT and Zaman K (1999) Oxidative stress as a mechanism of chronic
1984; Li and Hidalgo, 1996; Tsuji and Tamai, 1996; Kitagawa
cadmium-induced hepatotoxicity and renal toxicity and protection by antioxidants.
et al., 1999; Guo et al., 1999). In this study, both JBP923 and
Toxicol Appl Pharmacol 154:256 –263.
Shiffman ML, Hofmann CM, Contos MJ, Luketic VA, Sanyal AJ, Sterling RK,
JBP485 inhibited the uptake of Gly-Sar, a typical substrate
Ferreira-Gonzalez A, Mills AS and Garret C (1999) A randomized, controlled trial
of oligopeptide transporter, by intestinal BBMVs in a concen-
of maintenance interferon therapy for patients with chronic hepatitis C virus and
tration-dependent manner (Fig. 5). This suggests the possi-
persistent viremia. Gastroenterology 117:1164 –1172.
Takeda S, Ishihara K, Wakui Y, Amagaya S, Maruno M, Asao T and Kobashi K
bility that these small dipeptides are recognized by the trans-
(1996) Bioavailability study of glycyrrhetic acid after oral administration of gly-
porter, resulting in its rapid and complete absorption.
cyrrhizin in rats; relevance to the intestinal bacterial hydrolysis. J Pharm Phar- macol 48:902–905.
Further studies are needed to identify the transport sys-
Tsuji A and Tamai I (1996) Carrier-mediated intestinal transport of drugs. PharmRes 13:963–977.
Wang Z, Kurosaki Y, Nakayama T and Kimura T (1994) Mechanism of gastrointes-
Here we reported two novel dipeptide compounds that can
tinal absorption of glycyrrhizin in rats. Biol Pharm Bull 17:1399 –1403.
repair liver function after both i.v. and oral administrations.
Yagi A, Nagao, Okamura N, Ishizu T, Itoh H and Shida T (1998) Effect of cyclo(trans-
4-L-hydroxyprolyl-L-serine) from hydrolysate of human placenta on baby hamster
These compounds can also exert an antihepatitis effect di-
kidney -21/C-13 cells. Nat Med 52:156 –159.
rectly on cultured hepatocytes. It should also be noted that
Yi H, Nakashima I and Isobe K (1996) Enhancement of nitric oxide production from
activated macrophages by glycyrrhizin. Am J Chin Med 24:271–278.
glycyrrhizin has been reported to exert many types of biolog-ical activity, including antioxidant effect, anti-apoptosis ac-
Send reprint requests to: Professor Yuichi Sugiyama, Ph.D., Graduate
tion, and enhancement of nitric oxide production from acti-
School of Pharmaceutical Sciences, University of Tokyo, 7-3-1 Hongo, unkyo-ku, Tokyo 113-0033, Japan. E-mail: [email protected]
vated macrophages, although it is still unknown which of
CHILDHOOD DISINTEGRATIVE DISORDER: A CASE STUDY Sujoy Kumar Makar Audiologist and Speech-Language Pathologist Pamela Samaddar Audiologist and Speech-Language Pathologist Sharon Victoria Rout Internee AYJNIHH (ERC) KOLKATA -90 Abstract:- Introduction CDD is a developmental disorder that resembles autism, characterized by at least two years of nor
High Deductible Health Plan Preventive Drug List Effective January 1, 2013 The medications that qualify for the preventive drug list are outlined below by drug classification. Wellmark Drug CARDIOVASCULAR MEDICATIONS List Tier Generic name Brand Name* CALCIUM ANTAGONISTS nifedipine extended-release – ADALAT CC diltiazem – CARDIZEM/LA/CD, DILACOR XR nifedipine
 Dipeptides Orally Exhibiting Antihepatitis Activities
Dipeptides Orally Exhibiting Antihepatitis Activities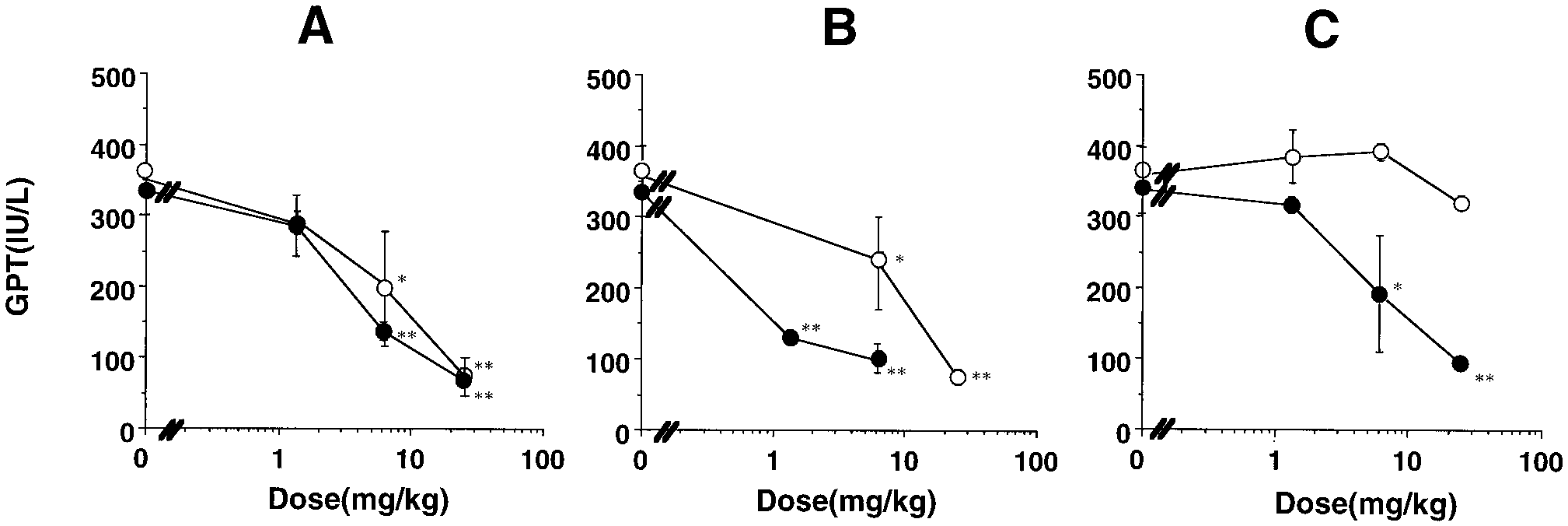
 Dipeptides Orally Exhibiting Antihepatitis Activities
Dipeptides Orally Exhibiting Antihepatitis Activities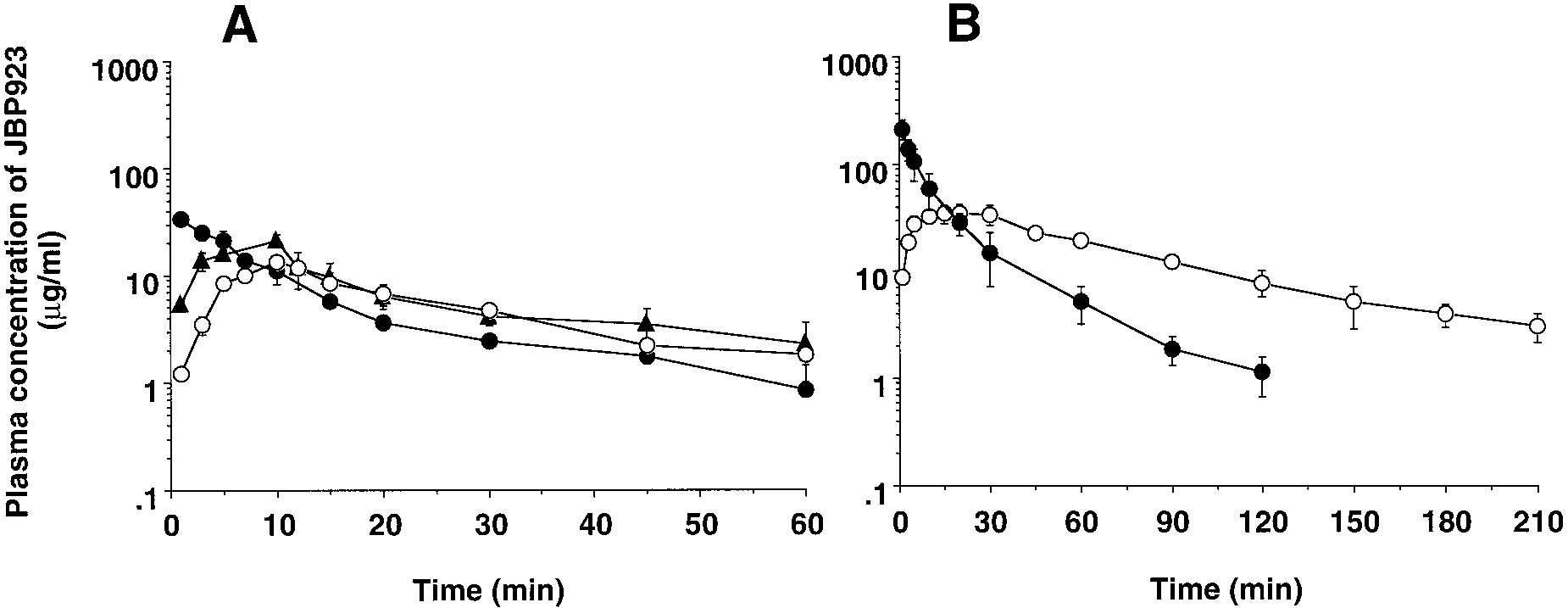
 Liu et al.
Liu et al.