Oxidative Coupling of 17 -Estradiol: Inventory of Oligomer Products and Configuration Assignment of Atropoisomeric C4-Linked Biphenyl-Type Dimers and Trimers
Alessandro Pezzella,* Liliana Lista, Alessandra Napolitano, and Marco d’Ischia
Department of Organic Chemistry and Biochemistry, University of Naples “Federico II” ComplessoUniversitario Monte S. Angelo, Via Cinthia 4, I-80126 Naples, Italy
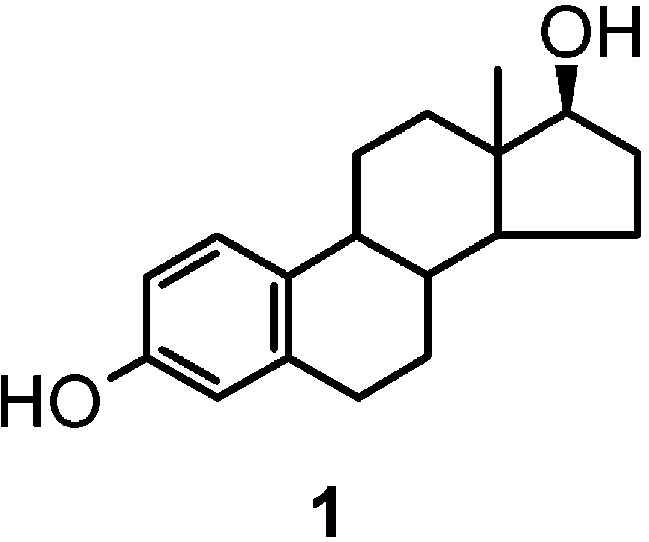
The oxidation chemistry of 17 -estradiol (1) is of central relevance to the nongenomic effects of estrogens and offers valuable prospects in the search for novel steroidal scaffolds of academic and industrial interest. Herein, we report the results of a detailed investigation into the nature of the oligomer products formed by phenolic oxidation of 1. Of the oxidants tested, the peroxidase/H2O2 system proved to be the most effective in inducing conversion of 1 to a complex mixture of oligomer species. Repeated chromatographic fractionation followed by extensive 2D NMR and mass spectrometric analysis allowed identification of a series of phenolic coupling products comprising, besides the C2-symmetric dimers 2 and 3, a 2,4′ dimer (4), two O-linked dimers (5, 6), and the novel trimers 7-9. All 4-linked biphenyl-type oligomers, i.e., 3 and 7-9, occurred as couples of atropoisomers, reflecting steric hindrance at biphenyl linkages. For all atropoisomers, absolute configuration was established by the exciton chirality method and the interconversion energy was determined by dynamic NMR. These results provide the first systematic inventory of oxidative coupling products of 1 and lay the foundation for future studies aimed to develop novel estrogen derivatives based on oligomeric scaffolds. Introduction
by, their potent antioxidant5 and free radical scavengingcapacity.6 In view of that, a detailed elucidation of the
17 -Estradiol (1) and structurally related estrogens
structural modifications suffered by 1 in oxidative set-
possess both carcinogenic1 and neuroprotective2 proper-
tings is central for the understanding of the nongenomic
ties that have been attributed to the inherent susceptibil-
effects of estrogens. In addition, beyond the specific
ity of the phenolic A-ring to enzymatic or chemical
relevance to the steroid sector, the oxidation of estrogen
oxidation.3 In particular, the involvement of 1 in breast
compounds represents an attractive research issue be-
and other human cancers would be the result of metabolic
cause of its potential as convenient entry to complex
conversion to the 2- and 4-hydroxy derivatives, termed
functionalized scaffolds of academic and industrial inter-
the catechol estrogens, and the corresponding o-quinones,
est, e.g. in asymmetric synthesis7 and supramolecular
which can induce the critical initiation step of tumori-
chemistry,8 in the quest for innovative lead compounds
genesis via adduct formation with DNA and depurination
in anticancer therapy,9 or for liquid crystal prepara-
processes.4 On the other hand, the neuroprotective action
tions,10 where 1 and related compounds are commonly
of estrogens would be related to, or at least complemented
(5) Behl, C.; Skutella, T.; Lezoualc’h, F.; Post, A.; Widmann, M.;
* To whom correspondence should be addressed. Fax: +39-081-
Newton, C. J.; Holsboer F. Mol. Pharmacol. 1997, 51, 535-541.
(6) (a) Dhandapani, K. M.; Brann, D. W. Biol. Reprod. 2002, 67,
(1) Yager, J. D. J. Natl. Cancer Inst. Monogr. 2000, 27, 67-73.
1379-1385. (b) Prokai, L.; Prokai-Tatrai, K.; Perjesi, P.; Zharikova,
Yager, J. D.; Liehr, J. G. Annu. Rev. Pharmacol. Toxicol. 1996, 36,
A. D.; Perez, E. J.; Liu, R.; Simpkins, J. W. Proc. Natl. Acad. Sci. U.S. A. 2003, 100, 11741-11746.
(2) (a) Bisagno, V.; Bowman, R. E.; Luine, V. E. Endocrine 2003,
(7) (a) Kaufmann, M. D.; Grieco, P. A.; Bougie, D. W. J. Am. Chem. 21, 33-41. (b) Kajta, M.; Beyer, C. Endocrine 2003, 21, 3-9. (c) Picazo, Soc. 1993, 115, 11648-11649. (b) Enev, V. S.; Ewers, Ch. L. J.; Harre,
O.; Azcoitia, I.; Garcia-Segura, L. M. Brain Res. 2003, 990, 20-27. (d)
M.; Nickisch, K.; Mohr, J. T. J. Org. Chem. 1997, 60, 364-369. (c)
Czlonkowska, A.; Ciesielska, A.; Joniec, I. Med. Sci. Monit. 2003, 9,
Strickler, R. R.; Schwan, A. L. Tetrahedron: Asymmetry 1999, 10,
247-256. (e) Moosmann, B.; Behl, C. Proc. Natl. Acad. Sci. U.S.A. 1999,
4065-4069. (d) Soulivong, D.; Matt, D.; Ziessel, R. J. Chem. Soc., Chem. Commun. 1999, 393-394. (e) Kostova, K.; Genov, M.; Philipova, V.
(3) (a) Bolton, J. L.; Yu, L.; Thatcher, G. R. Methods Enzymol. 2004, Tetrahedron: Asymmetry 2000, 11, 3253-3256. 378, 110-123. (b) Green, P. S.; Yang, S. H.; Simpkins, J. W. Novartis
(8) (a) Davis, A. P.; Joos, J.-B. Coord. Chem. Rev. 2003, 240, 143- Found. Symp. 2000, 230, 202-220.
156. (b) de Jong, M. R.; Knegtel, R. M. A.; Grootenhuis, P. D. J.;
(4) (a) Cavalieri, E. L.; Rogan, E. G.; Chakravarti, D. Cell Mol. Life
Huskens, J.; Reinhoudt, D. N. Angew. Chem., Int. Ed. Engl. 2002, 41, Sci. 2002, 59, 665-681. (b) Convert, O.; Van Aerden, C.; Debrauwer,
1004-1008. (c) Smith, D. K.; Diederich, F.; Zingg, A. Math. Phys. Sci.
L.; Rathahao, E.; Molines, H.; Fournier, F.; Tabet, J.-C.; Paris, A. Chem.1999, 527, 261-272. Res. Toxicol. 2002, 15, 754-764. (d) Markushin, Y.; Zhong, W.;
(9) (a) Lakhani, N. J.; Sarkar, M. A.; Venitz, J.; Figg, W. D.
Cavalieri, E. L.; Rogan, E. G.; Small, G. J.; Yeung, E. S.; Jankowiak
Pharmacotherapy. 2003, 23, 165-172. (b) Schumacher, G.; Neuhaus,
R. Chem. Res. Toxicol. 2003, 16, 1107-1117.
P. J. Cancer Res. Clin. Oncol. 2001, 7, 405-410
10.1021/jo0492665 CCC: $27.50 2004 American Chemical Society
J. Org. Chem. 2004, 69, 5652-5659 Oxidative Coupling of 17 -Estradiol
employed. The close bearing on the field of phenolic
By contrast, a substantial substrate consumption was
coupling, which is central to several areas of organic
observed with the peroxidase/H2O2 system, with forma-
chemistry, further warrants exploration of the oxidation
tion of a number of products whose chromatographic and
spectral properties were suggestive of oligomer species.
Yet, despite the many prospects offered by oxidative
The mechanistic background provided by the extensive
manipulation of estrogens, current knowledge in the field
literature on peroxidase-catalyzed oxidation of phenols16
is surprisingly limited. The only known oxidation prod-
and the occurrence of peroxidase in mammalian tissues
ucts include, besides the catechol estrogens, a 10 -
responsive to estrogen activity, such as uterus,17 war-
hydroxyestra-1,4-dien-3-one derivative arising by peracid-
ranted investigation of such reaction as a paradigm to
induced photooxygenation or oxidation by Fenton
detail the behavior of this estrogen on oxidation. Accord-
reagent,6b a series of benzylic oxidation species of estrone
ingly, we decided to embark on the isolation and detailed
methyl ether,11 and two dimers obtained by chemical and
characterization of the products formed by peroxidase/
enzymatic oxidation of 1, namely, the symmetric 2,2′ and
H2O2 oxidation of 1. As the medium, phosphate buffer at
4,4′ dimers.12 Other studies have appeared reporting
pH 7.4 with little methanol to favor estrogen solubiliza-
formation of oligomer species by oxidation of 1, but their
tion was preferably used. Two-phase systems, e.g. water-
characterization relied only on evaluation of chemical
ethyl acetate,12 proved of limited utility and were not
physical properties.13 More recently, a convenient syn-
thetic access to O-linked dimers of 1 was reported14 in
In a typical preparative scale reaction, 1 at 0.3 mM
the frame of a study of the NADPH-dependent metabo-
concentration was allowed to react with peroxidase (1
lism of 1 by human liver microsomes and cytochrome
U/mL) and hydrogen peroxide (2 mol equiv). After 60 min,
P450 enzymes. These latter studies and the vast body of
with >98% substrate consumption, the mixture was
literature on the oxidative coupling of phenols15 suggest
extracted with ethyl acetate following careful acidification
that oxidative conversion of 1 and related estrogens in
to pH 5.0. PLC fractionation afforded seven main chro-
vivo can lead to an array of oligomeric products, yet their
nature and biological properties have remained so far
0.22, and 0.10 (eluant A) designated A-G, in that order.
poorly elucidated. This study was therefore concerned
Of these, only fraction D consisted of a single species pure
with an investigation of the reaction behavior of 1 with
enough for spectroscopic analysis, whereas fractions A,
various oxidizing systems. Specific goals were to gain a
B, and D-F required further fractionation. The most
systematic insight into the modes of oxidative coupling
polar band G was made of chromatographically ill-defined
of 1 and to provide a detailed structural characterization
products, and their identity was not investigated.
of the oligomer products for future in vitro and in vivo
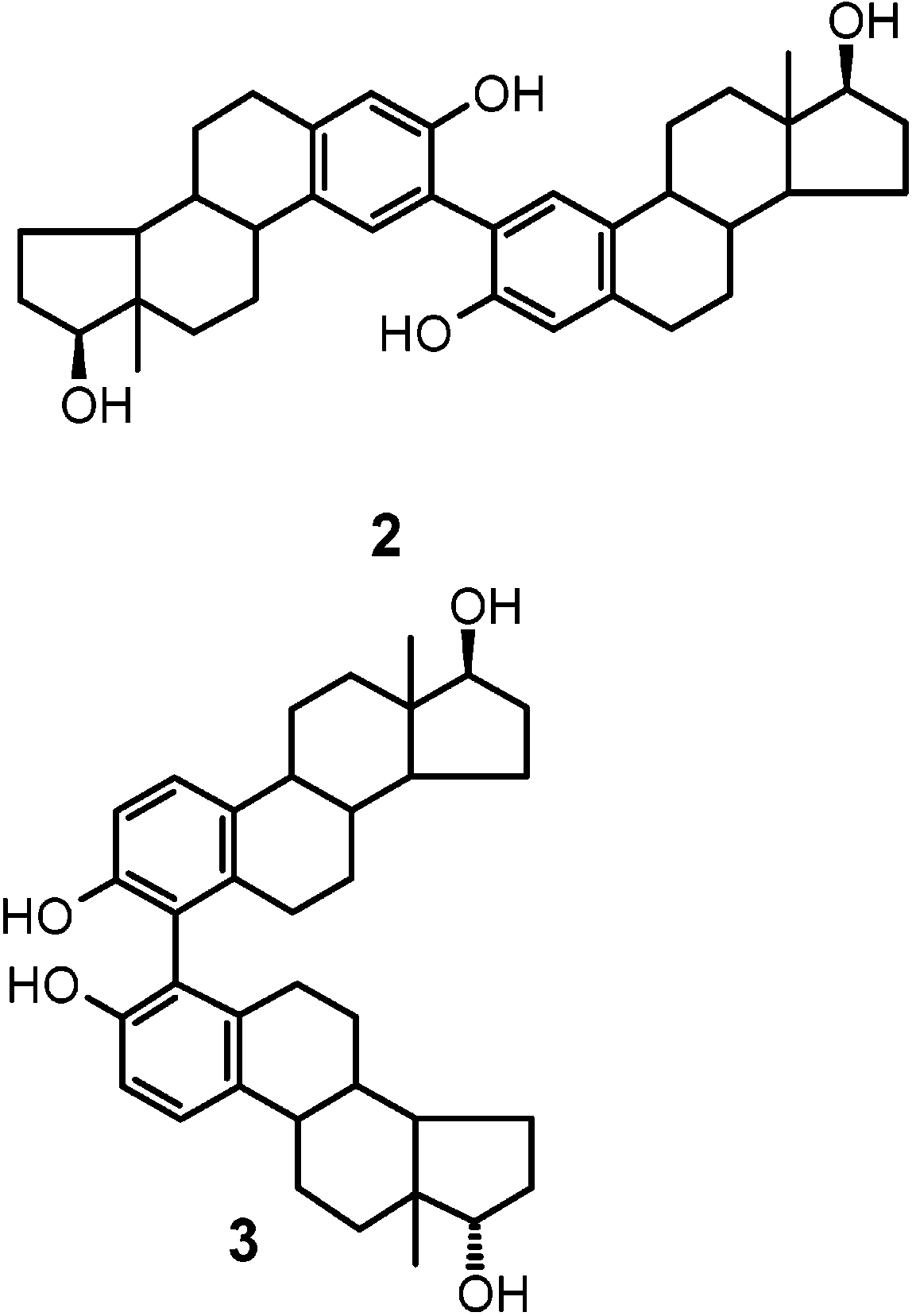
Spectral data (1H and 13C NMR) of the product from
band D were in agreement with a C2-symmetric dimer (molecular ion peak at m/z 542). Homo- and hetero- nuclear correlation experiments allowed straightforward formulation of the product as 2. Results and Discussion
In preliminary experiments, the ability of various
chemical and enzymatic oxidants to induce conversion of 1 to oligomer products was briefly investigated under different reaction conditions. With the chemical oxidants tested, i.e., persulfate, ferricyanide, and ceric ammonium nitrate, little or no substrate conversion was observed (HPLC and TLC evidence) in aqueous buffers or biphasic media in a broad range of pH values. With ferricyanide, slow substrate consumption was obtained only in 0.1 M NaOH, as previously reported.12
(10) (a) Brandenburger, F.; Matthes, B.; Seifert, K.; Strohriegl, P. Liq. Cryst. 2001, 28, 1035-1039. (b) McHugh, C.; Tomlin, D. W.; Bunning, T. J. Macromol. Chem. Phys. 1997, 198, 2387-2395.
(11) Modica, E.; Bombieri, G.; Colombo, D.; Marchini, N.; Ronchetti,
F.; Scala, A.; Toma, L. Eur. J. Org. Chem. 2003, 2964-2971.
Chromatographic band A consisted of an intimate
(12) Ius, A.; Meroni, G.; Ferrara, L. J. Steroid Biochem. 1977, 8,
mixture of two closely related species which could be
(13) (a) Norymberski, J. K. FEBS Lett. 1977, 76, 231-234. (b)
Norymberski, J. K. FEBS Lett. 1978, 86, 163-166. (c) Norymberski,
(16) (a) Uyama, H.; Kobayashi, S. Curr. Org. Chem. 2003, 7, 1387-
J. K. J. Steroid Biochem. Mol. Biol. 1991, 39, 73-81.
1397. (b) Uyama, H.; Kobayashi, S. J. Mol. Catal. B: Enzymol. 2002,
(14) Lee, A. J.; Sowell, J. W.; Cotham, W. E.; Zhu, B. T. Steroids19-20, 117-127. (c) Adam, W.; Lazarus, M.; Saha-Moller, C. R.;
2004, 69, 61-65.
Weichold, O.; Hoch, U.; Haring, D.; Schreier, P. Adv. Biochem. Eng./
(15) (a) Kobayashi, S.; Higashimura, H. Prog. Polym. Sci. 2003, 28, Biotechnol. 1999, 63 (Biotransformations), 73-108.
1015-1048. (b) Schmid, A.; Hollmann, F.; Buehler, B. Enzyme Cataly-
(17) (a) Jellinck, P. H.; McNabb, T.; Cleveland, S.; Lyttle, C. R. Adv.sis in Organic Synthesis, 2nd ed.; Drauz, K., Waldmann, H., Eds.;
Enzyme Regul. 1976, 14, 447-465. (b) O’Brien, P. J. Chem. Biol. Interact. 2000, 129, 113-139. J. Org. Chem, Vol. 69, No. 17, 2004 5653
separated by preparative HPLC. The products showed
TABLE 1. Selected NMR Data for Compounds 5 and 6
nearly identical 1H NMR spectra featuring in the aro-
matic region two doublets (J ) 8.8 Hz) at around δ 7.3
2-symmetric 4,4′ dimer 3. On
this basis, the two products were regarded as atropoiso-
mers arising by a restricted rotation around the sterically
crowded biphenyl 4,4′ linkage. No appreciable intercon-
version of the rotational isomers was observed by heating
to 110 °C at which temperature the products began to
decompose significantly. This implies that the activation
energy barrier is greater than 22.5 kcal mol-1.
The constituents of chromatographic band C as purified
by HPTLC displayed very similar 1H NMR spectra
showing in the aromatic region two singlets at about δ
7.0 and 6.8 and two doublets (J ) 8.4 Hz) at about δ 7.3and 6.9, suggesting two atropoisomers of a 2,4′-linked
showed very close proton spectra and were regarded as
dimer (4). This view was confirmed by dynamic 1H NMR
atropoisomers. Both displayed in the aromatic region an
experiments. Line shape analysis at a temperature
ABX spin system and three singlets around δ 6.7, 6.9,
around coalescence allowed calculation of the mean
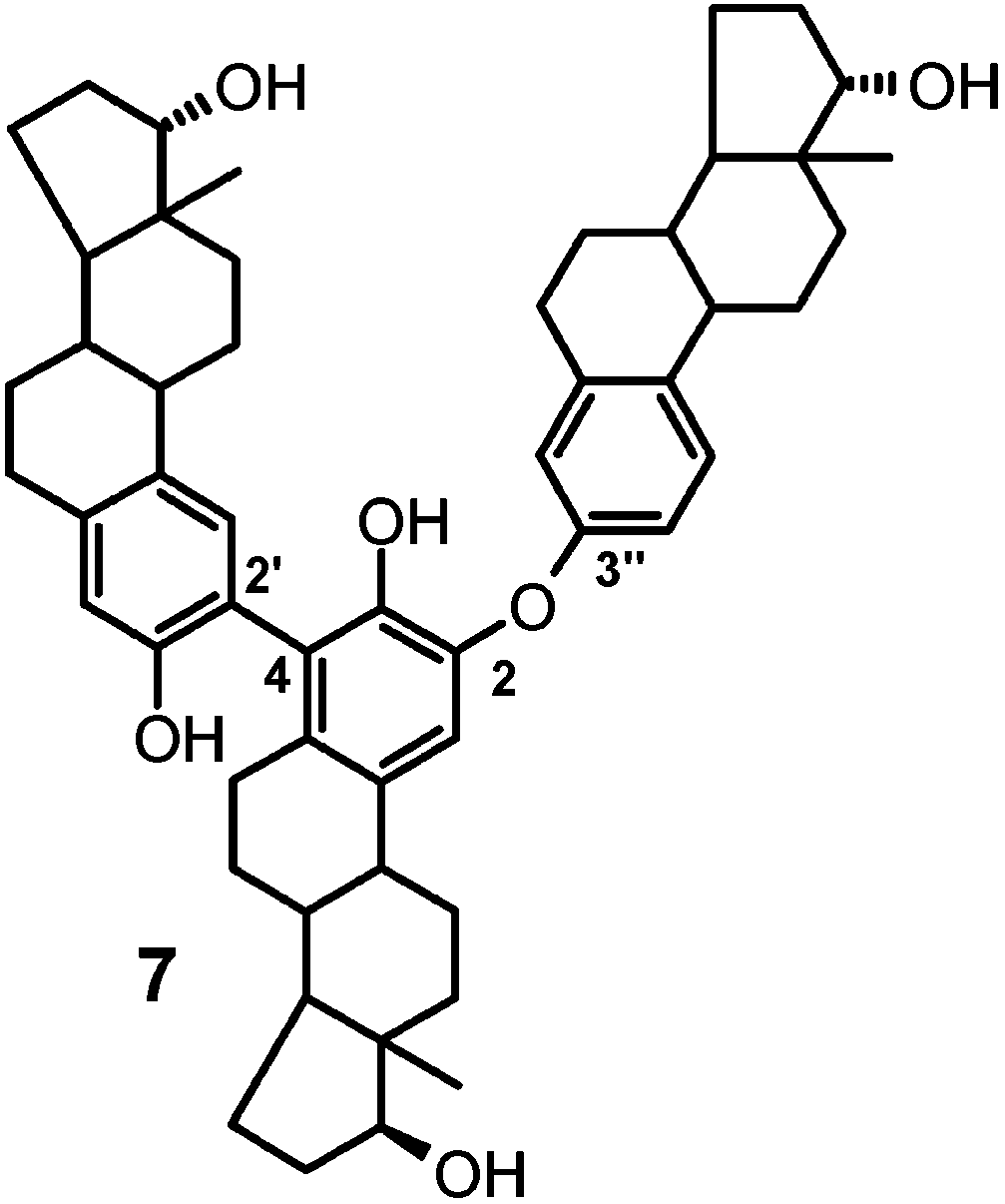
and 7.0, consistent with a trimer in which a central
lifetime of the atropoisomers, and a free energy of
estradiol unit is linked to the 2-position of an outer
activation of 21.5 ( 0.5 kcal mol-1 was determined by
moiety and to the oxygen of the other unit. On the basis
of the HMBC correlation data, it was possible to assign the signals at δ 7.04 and 6.75, for the faster HPLC eluted compound, to the H-1 and H-4 protons of the same estradiol unit. The shielding effect caused by the oxygen bridge, observed also in dimers 5 and 6, and the presence of a weak but well discernible cross-peak between the proton resonance at δ 7.04 and a substituted C-4 carbon resonance at δ 124.3 (see Table 2) allowed straightfor- ward assignment of the former signal to the H-1 proton of the central unit leading eventually to assign the
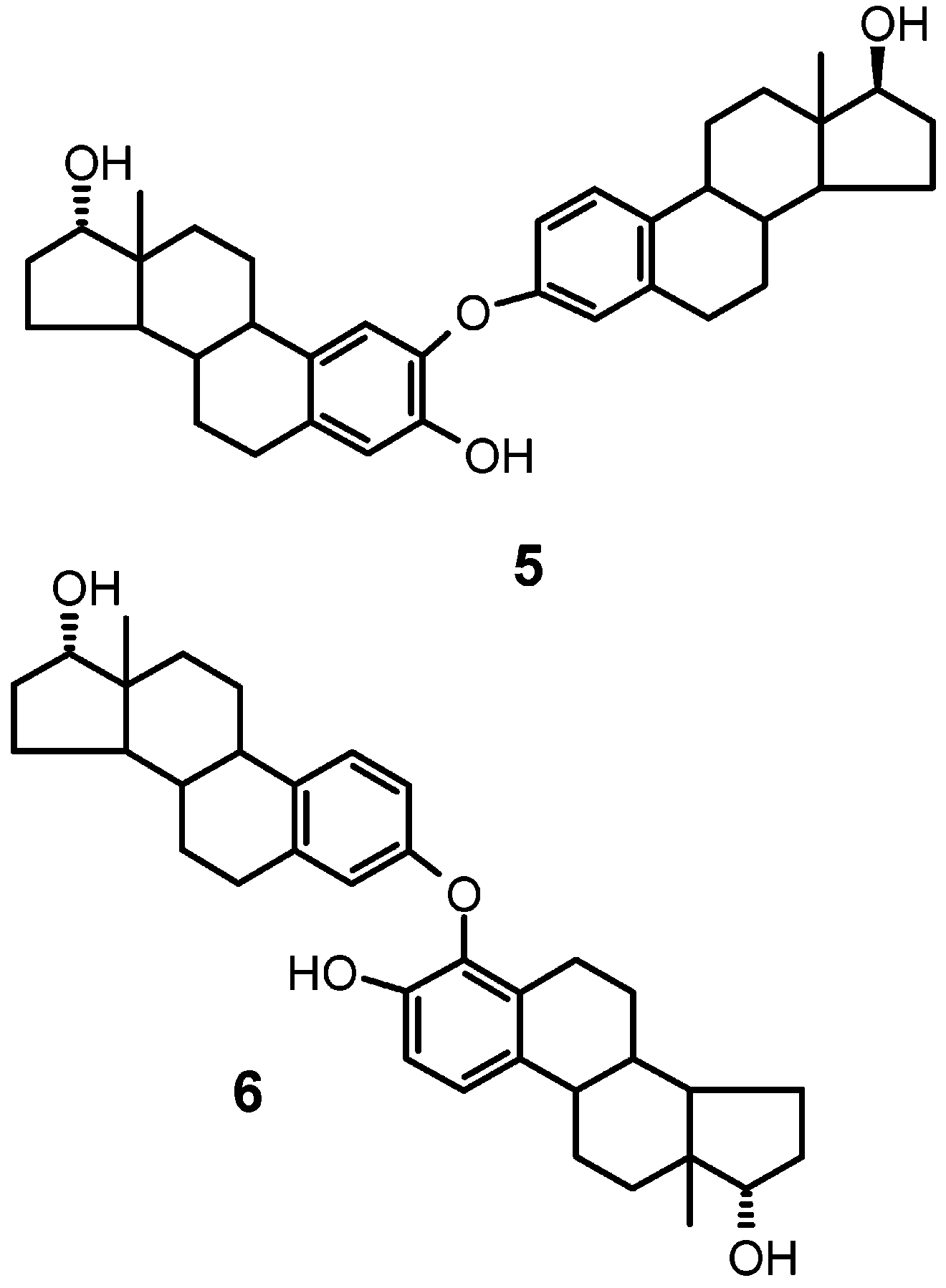
The two components of chromatographic band B were
trimers structure 7. In these, the atropoisomerism is
separated by preparative HPLC. The mass and NMR
apparently the result of the restricted rotation around
data led to straightforward formulation of the compounds
the 2,4′ linkage. NMR line shape analysis around the
as the O-linked dimers 5 and 6.14 Extensive 2D proton-
coalescence temperature allowed calculation of a free
proton and proton-carbon correlation experiments al-
energy of activation of 20.9 ( 0.4 kcal mol-1.
lowed complete assignment of the aromatic resonances(see Table 1).
Of the four main HPLC-separable products in band F,
those eluting at 24 and 80 min (eluant II) interchangedon heating, suggesting again an atropoisomer relation-ship, whereas those eluting at 34 and 37 min (eluant II)were not affected by heating to 100 °C.
The aromatic region of the 1H NMR spectrum of the
products eluted at 24 and 80 min displayed five singlets,a feature which was compatible with the trimeric struc-
All products from chromatographic bands E and F
ture 8. Complete assignment of the proton and carbon
exhibited molecular ion peaks at m/z 812 in the EI-MS
signals in the aromatic region was achieved on the basis
spectrum indicating trimeric structures. Products from
of the data of the correlation experiments. In particular,
band E as obtained in pure form by HPLC separation
in the case of the slower eluting isomer (8b), the singlets 5654 J. Org. Chem., Vol. 69, No. 17, 2004 Oxidative Coupling of 17 -EstradiolTABLE 2. Selected 1H-13C NMR Correlation Data for TABLE 3. Selected 1H-13C NMR Correlation Data for Compound 7a18 Compound 8b18 a Numbering as shown in formula 7. TABLE 4. Selected 1H-13C NMR Correlation Data for Compound 9b18
at δ 7.21 and δ 6.76 were assigned to H-1 and H-4 protons
of the same unit, respectively, on the basis of the 3J and
2J long-range contacts exhibited with C-3 and C-2 at δ
151.1 and 123.0, respectively. A 3J contact between the
latter carbon and the H-1′ proton at δ 7.32 of another
unit provided support to the 2,2′ linkage between two of
the trimer units (see Table 3). The free energy activation
for the interconversion was calculated as 21.3 ( 0.5 kcal
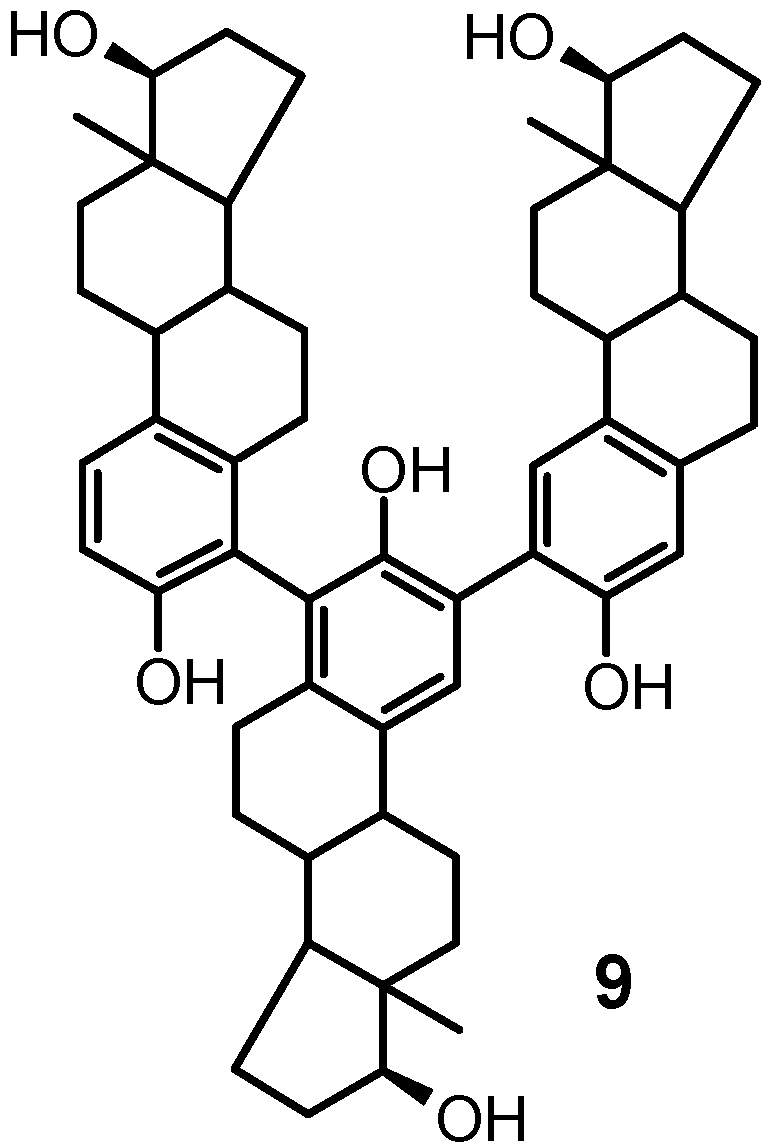
was compatible with either of the two trimeric structures in which the estradiol units were linked through the 2,4′: 2′,4′′ or the 2,2′:4′,4′′ positions. The lack of appreciable interconversion on heating, observed also for the atro- poisomers of the 4,4′ dimer 3, strongly argued in favor
The 1H and 13C spectra of the other two constituents
of structure 9. This assignment was confirmed by analy-
of band F were likewise very similar. The aromatic
sis of the proton-carbon correlation spectra showing
regions of the proton spectra diplayed three singlets and
contacts matching those of the 2,2′ subunit of trimer 8
two doublets (J ) 8.4 Hz), a pattern of resonance that
Interestingly, when the oxidation of 1 was carried out
with the substrate at 0.3 µM concentration, that is, at a concentration close to physiological values, substrate consumption was >95% at 1 h and the main reaction products were the dimers 2 and 4, whereas the O-linked dimers 5 and 6 were formed only in very small amounts and dimer 3 and trimers 7-9 were below detection limits.
For comparative purposes, the oxidation of 1 with
manganese dioxide in chloroform was briefly investi- gated. Under these conditions, a smooth oxidation of 1 (>99% consumption after 20 h) was observed with formation of dimers 3 and 4 as main species (about 30% overall formation yields) but with no detectable 2, 5, and 6. The different product patterns obtained at lower J. Org. Chem, Vol. 69, No. 17, 2004 5655 FIGURE 1. CD spectra of compounds 3a (A), 4a (B), and 7a (C). substrate concentration, or using manganese dioxide in chloroform, suggest that the generation and mode of coupling of phenoxyl radicals is under the influence of several factors. For example, tenuous steric factors may become significant under high dilution conditions, thus accounting for the lack of formation of the relatively hindered dimer 3, whereas solvent effects may explain the prevalence of C-coupling products, i.e. dimers 3 and 4 in chloroform, also furnishing a suggestion for prepara- tive purposes requiring regiochemically more restricted products patterns.
The sterically hindered biphenyl linkage in 3, 4, and 7-9 represents a stereogenic element which adds to those already present in 1. For all isolated products featuring such structural system, configuration at the biphenyl linkage (and thus absolute stereochemistry) was estab- lished by the exciton chirality method on the basis of the Cotton effect associated with the phenolic transition 1La, whose vector nearly overlaps that joining the C10-C3-O centers.19 This transition is observed at around 220 nm in 1 in EtOH, but the formation of biphenyl linkages and the presence of other substituents, such as the O-linked unit in 7a,b, cause shift to longer wavelengths. The FIGURE 2. CD spectra of compounds 8a (A) and 9a (B).
relative directions of the 1La transition dipoles and of thebiphenyl bonds allowed assignment of positive screwness
all isolated products, whereas the Cotton effect at ca. 280
configuration (P) to those isomers exhibiting positive
nm (transition 1Lb) was less defined for nearly all
Cotton effect independently from the regiochemistry of
products, with the exception of those featuring 4,4′-
the biphenyl linkage. Indeed, geometry optimization of
the oligomer structures (MM+) showed that the dihedral
On this basis, the negative Cotton effect of the first
angle between the planes of the aromatic rings of the
HPLC eluted atropoisomer of 4 and 7 (i.e. 4a and 7a)
biphenyl system has an absolute value ranging from 43°
indicates a negative helical orientation of phenol transi-
22′ to 45° 15′ for 2,4′ linkages and from 90° 02′ to 94° 92′
tion moments that means an M molecular chirality, while
for 4,4′ linkages. On the basis of the angles between 1La
the first eluted isomer of 3 has the P configuration
transition vectors and the dihedral intersection line,
trigonometric calculations gave the range +37 to +77°
In the case of 8 (i.e. 2,2′:4′,2′′-triestradiol) and 9 (i.e.
for the angle between the dipole transition moments in
2,2′:4′,4′′-triestradiol) the first eluted isomers share the
the case of a positive dihedral angle. These values are
M configuration at the 4′,2′′ biphenyl linkage and at the
significantly smaller than 110°, which is the theoretical
4′,4′′ linkage, respectively (Figure 2 A,B).
zero point at which for polyphenyl systems featuring
Mechanistically, formation of oligomer products 3-9
right-handed screwness19c the sign of the exciton split of
by peroxidase/H2O2 promoted oxidation of 1 can be
CD Cotton effect changes from positive to negative,
interpreted as involving generation and coupling of
allowing straightforward molecular configuration assign-
phenoxyl radicals from 1. In the presence of H2O2, ferric
peroxidase (ground state) generates the ferryl π cation
The choice of 1La transition arises also from the clearly
(compound I) via two electron oxidation. Compound I can
defined monosignated Cotton effect at around 230 nm in
then be reduced to compound II, the ferryl form of theenzyme, which has higher oxidative equivalents than the
(18) “a” and “b” lettering denotes the faster and the slower HPLC
resting ferric form.17b Both compounds I and II can
oxidize the phenolic moiety of 1 to give the phenoxyl
(19) (a) Harada, N.; Nakanishi, K. Circular Dichroic Spectroscopy-Exciton Coupling in Organic Stereochemistry; University Science
Books: Mill Valley, CA, 1983. (b) Dong, J. G.; Akritopoulou-Zanze, I.;
From inspection of the SOMO and Mulliken spin
Guo, J.; Berova, N.; Nakanishi, K.; Harada, N. Enantiomer 1997, 2,
densities of the phenoxyl radical of 1 reported in a
397-409. (c) Hanazaki, I.; Akimoto, H. J. Am. Chem. Soc. 1972, 94, 4102-4106.
previous study,20 no appreciable difference was antici-
5656 J. Org. Chem., Vol. 69, No. 17, 2004 Oxidative Coupling of 17 -Estradiol
pated in the reactivity of 1 through the 2 and 4 positions,
absorbance values in the range 0.1-0.2 at 220 nm. 1H (13C)
in accord with experimental evidence. Coupling through
NMR spectra were recorded at 400.1 (100.6) MHz. 1H-1H
the oxygen center is clearly a reflection of the high spin
COSY, 1H-13C HMQC, and 1H-13C HMBC experiments were
density at this site, in conformity with the known
run at 400.1 MHz using standard pulse programs from theBruker library. For electron impact (EI-MS) and high resolu-
patterns of oxidative coupling of phenols.
tion (HR-MS) mass spectra samples were ionized with a 70eV beam and the source was taken at 180-280 °C. Conclusions
Analytical and preparative TLC analyses were performed
on F254 0.25 and 0.5 mm silica gel plates or high performance
The analytical and structural undertaking described
TLC (HPTLC) using 40:60 cyclohexanes-ethyl acetate (eluant
herein fills an important gap in the current knowledge
A) or 98:2 chloroform-methyl alcohol (eluant B).
of the oxidation chemistry of estrogens and, more in
Analytical and preparative HPLC was performed with an
general, of natural phenolic compounds. Highlights of
instrument equipped with a UV detector set at 280 nm.
this study include (a) the first isolation and complete
Octadecylsilane-coated columns, 4.6 × 250 mm or 22 × 250
characterization of trimeric steroids linked through C-C
mm, 5 µm particle size, were used for analytical or preparative
and C-O-C bonds, (b) the first example, to the best of
runs, respectively. Flow rates of 1 or 15 mL/min were used. Different isocratic and gradient elution conditions were used
our knowledge, of atropoisomerism in steroidal systems,
generated by steric hindrance to free rotation at 2,4′- and
acetonitrile (eluant II); 90:10 H2O-acetonitrile (solvent A),
4,4′-biphenyl linkages, and (c) the exploitation of peroxi-
acetonitrile (solvent B), 0-5 min 30% B, 5-30 min 30-55%
dase/H2O2 as an efficient and clean oxidizing system in
Oxidation of 1 by the Peroxidase/H2O2 System: Gen-
From the biomedical point of view, the present results
eral Procedure. To a solution of 1 (5 mg, 1.9 × 10-5 mol) in
offer an improved background to elucidate the chemical
methanol (5 mL) were added 0.1 M phosphate buffer, pH 7.4
nature and fate of the products derived from the anti-
(60 mL), and peroxidase (1 U/mL) sequentially. The mixture
oxidant and radical scavenging reactions or from oxida-
was then treated with hydrogen peroxide in aliquots (8 × 2.5
× 10-6 mol) every 10 min while being kept under stirring at
tive changes of the estrogens at sites of inflammation and
room temperature. At different time intervals the reaction was
active metabolic transformation. In the light of the
carefully acidified at pH 5.0 and extracted three times with
suggested role of 1 as OH radical scavenger, generation
ethyl acetate (3 × 60 mL). The combined organic layers were
of these oligomers may represent an alternative outcome
dried over sodium sulfate and analyzed by HPLC (eluant III)
of the radical scavenging action in addition to quinol
and TLC (eluant A). In other experiments the reaction was
formation.6b Oligomers 5 and 6 resemble the photodeg-
carried out as above with the substrate at 3 × 10-7 M
radation products of ethinyl estradiol,21 and their forma-
concentration using peroxidase (0.02 U/mg) and hydrogenperoxide (1 mol equiv)
tion by autoxidation and photodegradation of estradiol-
Oxidation of 1 by MnO
containing drugs can be predicted. C2. A solution of 1 (10 mg, 3.7 ×
10-5 mol) in chloroform (10 mL) was treated with MnO2 (64
bear considerable similarity to stereochemically related
mg, 8 × 10-4 mol) and kept overnight at room temperature.
products22 currently under scrutiny because of their
The solid was removed by centrifugation, and the mixture was
antiestrogenic activity and may represent attractive
taken to dryness, taken up in methanol, and analyzed by
prototypes/leads for the rational design of new bioactive
HPLC (eluant III) and TLC (eluant A). Isolation of Compounds 2-9. For preparative purposes,
Finally, atropoisomeric estradiol oligomers are analo-
reaction of 1 with peroxidase/H2O2 was run as described above
gous to para-polyaryls, which exhibit attractive structural
using 500 mg (1.84 × 10-3 mol) of the starting material at 3.0
×10-4 M concentration. After workup of the reaction mixture,
features, such as helicity, and other connected unusual
the residue obtained (480 mg) was fractionated by PLC (eluant
chemical-physical properties underlying a number of
A) to give seven fractions. Fraction A (15 mg, R )
A) was further purified by preparative HPLC (eluant II) to
The extension of the scope and utility of the oxidation
give pure 3a (5 mg, t )
8 min, eluant II, 1% yield) and 3b (5
chemistry of estrogens is currently a matter of concern
17 min, eluant II, 1% yield). Fraction B (25 mg, Rf
0.55 eluant A) was fractionated by PLC (eluant I) to give pure 5 (8 mg, t )
27 min, eluant II, 1.6% yield) and 6 (8 mg, tr
29 min, eluant II, 1.6% yield). Fraction C (15 mg, R ) 0.45
Experimental Section
eluant A) was purified by HPTLC (eluant B) to afford 4a (5 General Methods. 17 -Estradiol (1), manganese(IV) diox-
9 min, eluant II, 1% yield) and 4b (5 mg, tr
ide activated 5 µm (85%), and hydrogen peroxide (30% w/w
eluant II, 1% yield). Fraction D (20 mg, Rf
solution in water) were used as obtained. Horseradish peroxi-
consisted of pure 2 (tr
0.33, eluant A) was purified by preparative
2O2 oxidoreductase; EC 1.11.1.7) type II and
mushroom tyrosinase (EC 1.14.18.1) were used.
HPLC (eluant II) to afford 7a (3 mg, tr
UV spectra were performed using a diode array spectro-
0.6% yield) and 7b (3 mg, tr
photometer. CD spectra were taken on spectropolarimeter at
25 °C using solutions of the products in ethanol exhibiting
preparative HPLC (eluant II) to give four bands corresponding to pure 8a (3 mg, t )
24 min, eluant II,, 0.6% yield), 8b
80 min, eluant II, 0.6% yield), 9a (3 mg, tr
(20) Pezzella, A.; Manini, P.; Di Donato, P.; Boni, R.; Napolitano,
min, eluant II, 0.6% yield), and 9b (3 mg, t )
A.; Palumbo, A.; d’Ischia, M. Bioorg. Med. Chem. 2004, 12, 2927-2936.
II, 0.6% yield). Fraction G (35 mg, R )
(21) Segmuller, B. E.; Armstrong, B. L.; Dunphy, R.; Oyler, A. R. J.Pharm. Biomed. Anal. 2000, 23, 927-37.
found to consist of a complex pattern of species and was not
(22) (a) Rabouin, D.; Perron, V.; N’Zemba, B.; C-Gaudreault, R.;
Berube, G. Bioorg. Med. Chem. Lett. 2003, 13, 557-560 (b) Portoghese, 2,2′-Bis[estra-1,3,5(10)-trien-3,17 -diol] (2). UV [λmax (CH3-
P. S. J. Med. Chem. 1992, 35, 1927-1937.
OH)]: 288 nm. 1H NMR (CD3OD), δ (ppm): 0.75 (s, 3H × 2),
(23) (a) Berresheim, A. J.; Mueller, M.; Muellen, K. Chem. Rev. 1999, 99, 1747-1785. (b) Martin, R. E.; Diederich, F. Angew. Chem., Int.
1.0-1.8 (m, 8H × 2), 1.9-2.1 (m, 4H × 2), 2.15 (m, 1H × 2),
Ed. Engl. 1999, 38, 1351-1377.
2.25 (m, 1H × 2), 2.85 (m, 1H × 2), 3.64 (m, 1H × 2), 6.31 (s,
J. Org. Chem, Vol. 69, No. 17, 2004 5657
1H × 2), 7.14 (s, 1H × 2). 13C NMR (CD3OD), δ (ppm): 12.6 (2
139.7 (C), 147.4 (C), 155.7 (C). EI/MS (m/z): 542, [M]+.
× CH3), 24.1 (2 × CH2), 29.3 (2 × CH2), 31.5 (2 × CH2), 31.8
HREIMS (m/z): calcd mass for C36H46O4, 542.3396; found,
(4 × CH2), 38.8 (2 × CH2), 41.1 (2 × CH), 45.1 (2 × C), 46.1 (2
× CH), 52.1 (2 × CH), 83.3 (2 × CH), 118.1 (2 × CH), 127.0 (2
2-[[(17 )-17-Hydroxy-19-norpregna-1,3,5(10)-trien-3-yl]-
× C), 130.3 (2 × CH), 134.7 (2 × C), 139.1 (2 × C), 153.0 (2 ×
oxy]-4,2′-bis[estra-1,3,5(10)-trien-3,17 -diol] (7a). UV [λmax
C). EI/MS (m/z): 542, [M]+. HREIMS (m/z): calcd mass for
(CH3OH)]: 288 nm. 1H NMR (CDCl3), δ (ppm): 0.78 (s, 3H),
C36H46O4, 542.3396; found, 542.3401.
0.79 (s, 3H) 0.80 (s, 3H), 1.1-1.7 (m, 21H), 1.70-1.85 (m, 3H),
4,4′-Bis[estra-1,3,5(10)-trien-3,17 -diol] (3a). UV [λ
1.85-2.00 (m, 5H), 2.1-2.2 (m, 5H), 2.2-2.3 (m, 3H), 2.35 (m,
2H), 2.45 (m, 1H), 2.55 (m, 1H), 2.85 (m, 4H), 3.74 (m, 3H),
3OH)]: 288 nm. 1H NMR (CDCl3), δ (ppm): 0.80 (s, 3H ×
2), 1.1-1.7 (m, 8H × 2), 1.78 (m, 1H × 2), 1.95 (m, 1H × 2),
6.75 (s, 1H), 6.76 (d, J ) 2.4 Hz, 1H), 6.80 (dd, J ) 8.4, 2.4
2.11 (m, 1H × 2), 2.15-2.30 (m, 2H × 2), 2.30-2.40 (m, 2H ×
Hz, 1H), 6.98 (s, 1H), 7.04 (s, 1H), 7.23 (d, J ) 8.4, 1H). 13C
2), 3.73 (t, J ) 8.2 Hz, 1H × 2), 6. 87 (d, J ) 8.8 Hz, 1H × 2),
NMR (CDCl3), δ (ppm): 11.9 (CH3), 23.9 (CH2), 27.1 (CH2),
7.30 (d, J ) 8.8 Hz, 1H × 2). 13C NMR (CDCl
2), 27.9 (CH2), 30.5 (CH2), 31.4 (CH2), 31.7 (CH2), 37.5
(CH), 38.9 (C), 39.5 (C), 44.0 (CH), 44.8 (CH), 45.1 (CH), 50.9
3), 23.9 (2 × CH2), 27.1 (2 × CH2), 27.8 (2 × CH2),
(CH), 82.7 (CH), 115.4 (CH), 116.4 (CH), 117.8 (CH), 118.4
2), 31.4 (2 × CH2), 37.5 (2 × CH2), 39.1 (2 × CH),
44.0 (2 × C), 45.0 (2 × CH), 50.9 (2 × CH), 82.6 (2 × CH),
(CH), 119.8 (C), 124.3 (C), 127.4 (CH), 128.3 (CH), 132.3 (C),
113.6 (2 × CH), 119.9 (2 × C), 128.0 (2 × CH), 134.4 (2 × C),
133.6 (C), 133.9 (C), 134.3 (C), 136.0 (C), 139.3 (C), 142.2 (C),
137.8 (2 × C), 152.0 (2 × C). EI/MS (m/z): 542, [M]+. HREIMS
144.6 (C), 151.6 (C), 155.9 (C). EI/MS (m/z): 812, [M]+.
HREIMS (m/z): calcd mass for C54H68O6, 812.5016; found,
3b. UV [λmax (CH3OH)]: 288 nm. 1H NMR (CDCl3), δ (ppm): 7b. UV [λ
0.81 (s, 3H × 2), 1.1-1.7 (m, 8H × 2), 1.79 (m, 1H × 2), 1.99
(ppm): 0.77 (s, 3H), 0.78 (s, 3H) 0.79 (s, 3H), 1.1-1.7 (m, 24
(s, 1H × 2), 2.12 (m, 1H × 2), 2.15-2.30 (m, 2H × 2), 2.30-
H), 1.7-2.0 (m, 5H), 2.0-2.3 (m, 8H), 2.40 (m, 2H), 2.50 (m,
2.40 (m, 2H × 2), 3.74 (t, J ) 8.2 Hz, 1H × 2), 6. 86 (d, J ) 8.8
2H), 2.87 (m, 4H), 3.72 (m, 3H), 6.75 (d, J ) 2.4 Hz, 1H), 6.77
Hz, 1H × 2), 7.32 (d, J ) 8.8 Hz, 1H × 2). EI/MS
(s, 1H), 6.81 (dd, J ) 8.4, 2.4 Hz, 1H), 6.98 (s, 1H), 7.03 (s,
(m/z): 542, [M]+. HREIMS (m/z): calcd mass for C36H46O4,
1H), 7.24 (d, J ) 8.4, 1H). EI/MS (m/z): 812, [M]+. HREIMS
(m/z): calcd mass for C54H68O6, 812.5016; found, 812.5026. 2,4′-Bis[estra-1,3,5(10)-trien-3,17 -diol] (4a). UV [λmax 2,2′:4′,2”-Tris[estra-1,3,5(10)-trien-3,17 -diol] (8a). UV
(CH3OH)]: 288 nm. 1H NMR (CDCl3), δ (ppm): 0.79 (s, 6H),
[λmax (CH3OH)]: 288 nm. 1H NMR (CDCl3), δ (ppm): 0.78 (s,
1.1-1.8 (m, 16H), 1.9-2.0 (m, 4H), 2.10 (m, 2H), 2.25 (m, 2H),
3H), 0.80 (s, 6H), 1.1-1.9 (m, 24H), 1.9-2.0 (m, 6H), 2.1-2.4
2.32 (m, 2H), 2.50 (m, 2H), 2.85 (m, 2H), 3.73 (m, 2H), 6.77 (s,
(m, 10H), 2.42 (m, 2H), 2.90 (m, 3H), 3.73 (m, 3H), 6.76 (s,
1H), 6.85 (d, J ) 8.4 Hz, 1H), 7.00 (s, 1H), 7.29 (d, J ) 8.4 Hz,
1H), 6.79 (s, 1H), 7.04 (s, 1H), 7.20 (s, 1H), 7.32 (s, 1H).
1H). 13C NMR (CDCl3), δ (ppm): 11.9 (CH3), 23.9 (CH2), 27.3
EI/MS (m/z): 812, [M]+. HREIMS (m/z): calcd mass for
(CH2), 27.8 (CH2), 28.0 (CH2), 28.3 (CH2), 30.4 (CH2), 31.4
C54H68O6, 812.5016; found, 812.5005.
(CH2), 31.7 (CH2), 37.50 (CH2), 37.52 (CH2), 39.0 (CH), 39.5
8b. UV [λmax (CH3OH)]: 288 nm. 1H NMR (CDCl3), δ
(CH), 44.0 (C), 44.8 (CH), 45.0 (CH), 50.8 (CH), 83.3 (CH),
(ppm): 0.77 (s, 3H), 0.78 (s, 6H), 1.1-1.9 (m, 24H), 1.9-2.0
113.6 (CH), 116.7 (CH), 117.4 (C), 120.5 (C), 127.9 (CH), 128.3
(m, 5H), 2.0-2.2 (m, 5H), 2.2-2.5 (m, 8H), 2.90 (m, 3H), 3.73
(CH), 133.3 (C), 134.5 (C), 136.9 (C), 140.1 (C), 152.3 (C). EI/
(m, 3H), 6.76 (s, 1H), 6.78 (s, 1H), 7.04 (s, 1H), 7.21 (s, 1H),
MS (m/z): 542, [M]+. HREIMS (m/z): calcd mass for C36H46O4,
7.32 (s, 1H). 13C NMR (CDCl3), δ (ppm): 11.8 (CH3), 23.9 (CH2),
24.0 (CH2), 24.5 (CH2), 27.2 (CH2), 27.9 (CH2), 29.3 (CH2), 30.1
4b. UV [λmax (CH3OH)]: 288 nm. 1H NMR (CDCl3), δ
(CH2), 30.4 (CH2), 30.5 (CH2), 31.4 (CH2), 37.5 (CH2), 39.1 (CH),
(ppm): 0.78 (s, 6H), 1.1-1.8 (m, 16H), 1.9-2.1 (m, 4H), 2.1-
39.4 (CH), 39.6 (CH), 44.0 (C), 44.8 (CH), 45.1 (CH), 50.9 (CH),
2.3 (m, 4H), 2.3-2.5 (m, 4H), 2.92 (m, 2H), 3.72 (m, 2H), 6.78
82.7 (CH), 116.9 (CH), 118.2 (CH), 121.9 (C), 122.5 (C), 123.0
(s, 1H), 6.87 (d, J ) 8.4 Hz, 1H), 6.99 (s, 1H), 7.29 (d, J ) 8.4
(C), 128.6 (CH), 128.9 (CH), 130.0 (CH), 133.3 (C), 133.9 (C),
Hz, 1H). EI/MS (m/z): 542, [M]+. HREIMS: calcd mass for
137.4 (C), 138.3 (C), 140.0 (C), 150.9 (C), 151.1 (C). EI/MS
C36H46O4, 542.3396; found, 542.3401.
(m/z): [M]+. HREIMS (m/z): calcd mass for C54H68O6, 812.5016;
2-[[(17 )-17-Hydroxy-19-norpregna-1,3,5(10)-trien-3-yl]- oxy]estra-1,3,5(10)-trien-3,17 -diol (5). UV [λ 2,2′:4′,4”-Tris[estra-1,3,5(10)-trien-3,17 -diol] (9a). UV
max (CH3OH)]: 288 nm. 1H NMR (CDCl3), δ (ppm): 0.79 (s,
1.6 (m, 16H), 1.6-1.8 (m, 2H), 1.90 (m, 2H), 1.95 (m, 1H), 2.0-
3H), 0.80 (s, 3H), 0.81 (s, 3H), 1.2-1.8 (m, 24H), 1.8-2.0 (m,
2.2 (m, 5H), 2.32 (m, 1H), 2.83 (m, 3H), 3.71 (m, 2H), 6.70 (d,
6H), 2.0-2.2 (m, 4H), 2.2-2.4 (m, 8H), 2.89 (m, 3H), 3.74 (m,
J ) 2.4 Hz, 1H), 6.74 (s, 1H), 6.76 (dd, J ) 8.4, 2.4 Hz, 1H),
3H), 6.76 (s, 1H), 6.88 (d, J ) 8.4 Hz, 1H), 7.22 (s, 1H), 7.32
6.87 (s,1H), 7.22 (d, J ) 8.4 Hz, 1H). 13C NMR (CDCl
(d, J ) 8.4, 1H), 7.34 (s, 1H). EI/MS (m/z): [M]+. HREIMS
3), 23.9 (CH2), 27.0 (CH2), 27.1 (CH2), 27.9
2), 28.0 (CH2), 29.9 (CH2), 30.4 (CH2), 31.4 (CH2), 37.4
λmax (CH3OH)]: 288 nm. 1H NMR (CDCl3), δ
(ppm): 0.79 (s, 3H), 0.80 (s, 6H), 1.1-1.8 (m, 24H), 1.8-2.0
2), 37.5 (CH2), 39.40 (CH), 39.45 (CH), 44.0 (C), 44.8 (CH),
50.8 (CH), 82.6 (CH), 114.8 (CH), 116.7 (CH), 117.6 (CH), 117.7
(m, 5H), 2.1-2.2 (m, 5H), 2.2-2.5 (m, 8H), 2.8-2.9 (m, 3H),
(CH), 127.4 (CH), 133.7 (C), 134.2 (C) 135.8 (C), 139.3 (C), 141.5
3.73 (m, 3H), 6.76 (s, 1H), 6.89 (d, J ) 8.4 Hz, 1H), 7.22 (s,
(C), 146.2 (C), 156.0 (C). EI/MS (m/z): 542, [M]+. HREIMS
1H), 7.33 (d, J ) 8.4, 1H), 7.36 (s, 1H). 13C NMR (CDCl3), δ
3), 11.9 (CH3), 23.8 (CH2), 24.0 (CH2), 24.5
4-[[(17 )-17-Hydroxy-19-norpregna-1,3,5(10)-trien-3-yl]-
(CH2), 26.5 (CH2), 28.1 (CH2), 29.7 (CH2), 30.5 (CH2), 31.1
oxy]estra-1,3,5(10)-trien-3,17 -diol (6). UV [λ
(CH2), 31.4 (CH2), 37.5 (CH2), 38.6 (CH), 39.5 (CH), 42.1 (C),
44.0 (CH), 44.9 (CH), 48.8 (CH), 50.8 (CH), 82.7 (CH), 118.3
3), δ (ppm): 0.78 (s, 6H), 1.1-1.8 (m,
16H), 1.8-1.9 (m, 2H), 1.9-2.0 (m, 2H), 2.0-2.2 (m, 4H), 2.33
(CH), 118.6 (CH), 118.9 (C), 122.6 (C), 122.7 (C), 128.2 (CH),
(m, 2H), 2.40 (m, 1H), 2.73 (m, 1H), 2.81 (m, 2H), 3.73 (m,
130.1 (CH), 130.2 (CH), 132.6 (C), 134.3 (C), 136.3 (C), 136.5
2H), 6.60 (d, J ) 2.4 Hz, 1H), 6.62 (dd, J ) 8.4, 2.4 Hz, 1H),
(C), 138.2 (C), 148.0 (C), 151.5 (C), 152.1 (C). EI/MS (m/z): 812,
6.86 (d, J ) 8.4, 1H), 7.10 (d, J ) 8.4 Hz, 1H), 7.17 (d, J ) 8.4
[M]+. HREIMS (m/z): calcd mass for C54H68O6, 812.5016; found,
24.6 (CH2), 27.0 (CH2), 28.0 (CH2), 28.2 (CH2), 30.5 (CH2), 31.4(CH
Acknowledgment. This study was carried out in
2), 37.4 (CH2), 39.0 (CH), 39.5 (CH), 44.0 (C), 44.8 (CH),
50.8 (CH), 82.7 (CH), 112.8 (CH), 113.8 (CH), 115.6 (CH), 123.6
the frame of MIUR (“Neurosteroidi e loro modificazioni
(CH), 127.5 (CH), 131.5 (C), 134.4 (C), 135.5 (C), 139.4 (C),
ossidative e nitrosative nel sistema nervoso dei cefa-
5658 J. Org. Chem., Vol. 69, No. 17, 2004 Oxidative Coupling of 17 -Estradiol
lopodi”, PRIN 2002, “Sostanze naturali ed analoghi
Supporting Information Available: 1H NMR spectra
sintetici ad attivita` antitumorale”, PRIN 2003) and
or selected regions of compounds 2-9 and 1H-13C HMBC
Regione Campania projects (L.R. 5, a. 2002). We thank
spectra of compounds 3a, 4a, 5, 7a, 8a, and 9a. This
the “Centro Interdipartimentale di Metodologie Chimico-
material is available free of charge via the Internet at
Fisiche” (CIMCF, University of Naples Federico II) for
NMR and mass facilities. We thank Mrs. SilvanaCorsani for technical assistance. J. Org. Chem, Vol. 69, No. 17, 2004 5659
La intención de estas actividades es facilitar a los alumnos y a las alumnas la lectura de esta adaptación de La Celestina . Para ello, al principio y al final se incluyen unas fichas con preguntas de carácter general acerca de la obra, de la época o del autor. El resto de las fichas se compone de actividades específicas (de comprensión, análisis y expresión), relacionadas con el conten
The relentless, focused binding of FOSRENOL.1-4 For sustained phosphate control — meal after meal, day after day, week after week.1, 5, 6 FOSRENOL removes phosphate, removes phosphate, removes phosphate, removes pho Fosrenol* 500mg, 750mg and 1000mg Chewable be exercised in these patients, and monitoring of liver function may be required. oedema; pain; thirst; blood aluminium increased;
 Oxidative Coupling of 17 -Estradiol
employed. The close bearing on the field of phenolic
By contrast, a substantial substrate consumption was
coupling, which is central to several areas of organic
observed with the peroxidase/H2O2 system, with forma-
chemistry, further warrants exploration of the oxidation
tion of a number of products whose chromatographic and
spectral properties were suggestive of oligomer species.
Oxidative Coupling of 17 -Estradiol
employed. The close bearing on the field of phenolic
By contrast, a substantial substrate consumption was
coupling, which is central to several areas of organic
observed with the peroxidase/H2O2 system, with forma-
chemistry, further warrants exploration of the oxidation
tion of a number of products whose chromatographic and
spectral properties were suggestive of oligomer species.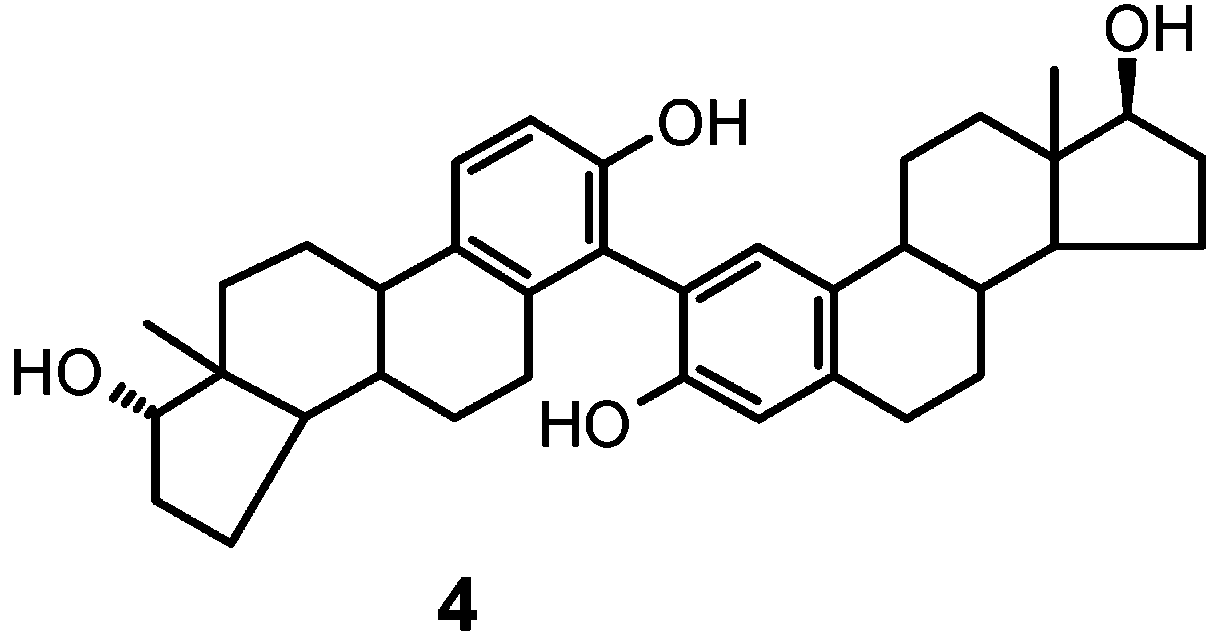
 separated by preparative HPLC. The products showed
TABLE 1. Selected NMR Data for Compounds 5 and 6
separated by preparative HPLC. The products showed
TABLE 1. Selected NMR Data for Compounds 5 and 6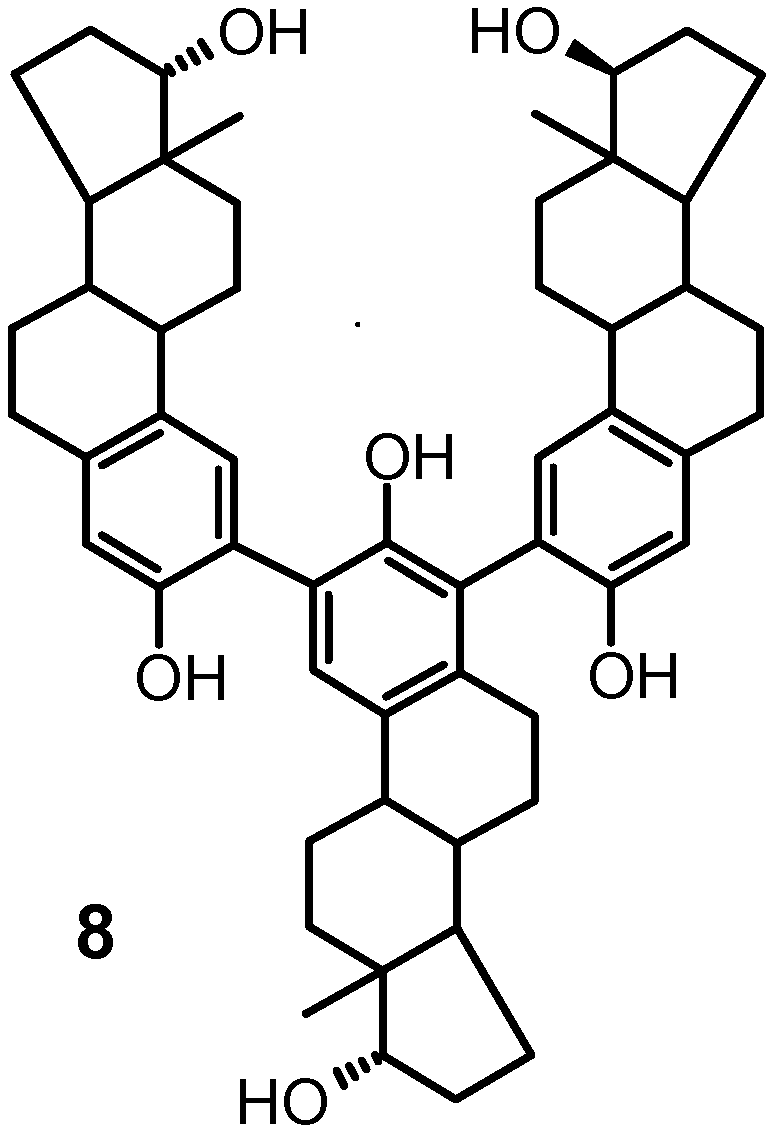
 Oxidative Coupling of 17 -Estradiol
TABLE 2. Selected 1H-13C NMR Correlation Data for
Oxidative Coupling of 17 -Estradiol
TABLE 2. Selected 1H-13C NMR Correlation Data for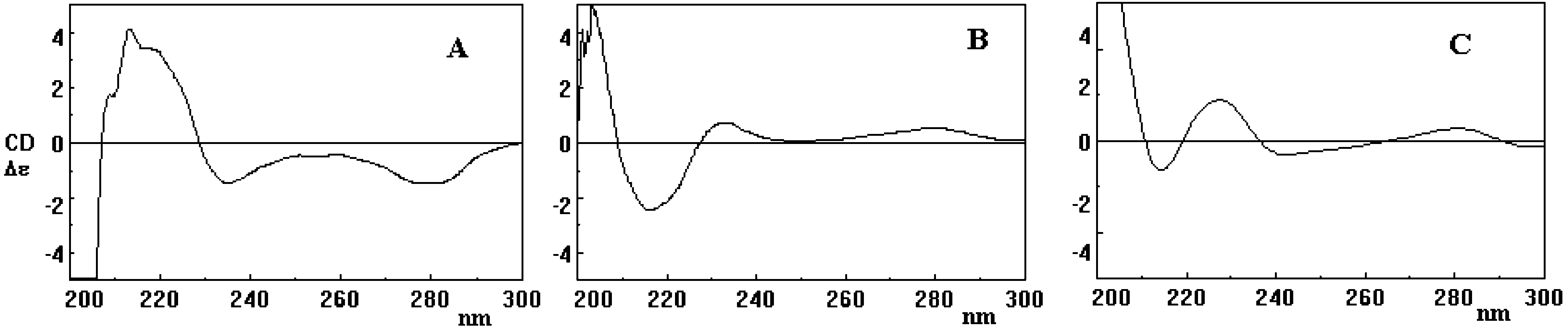
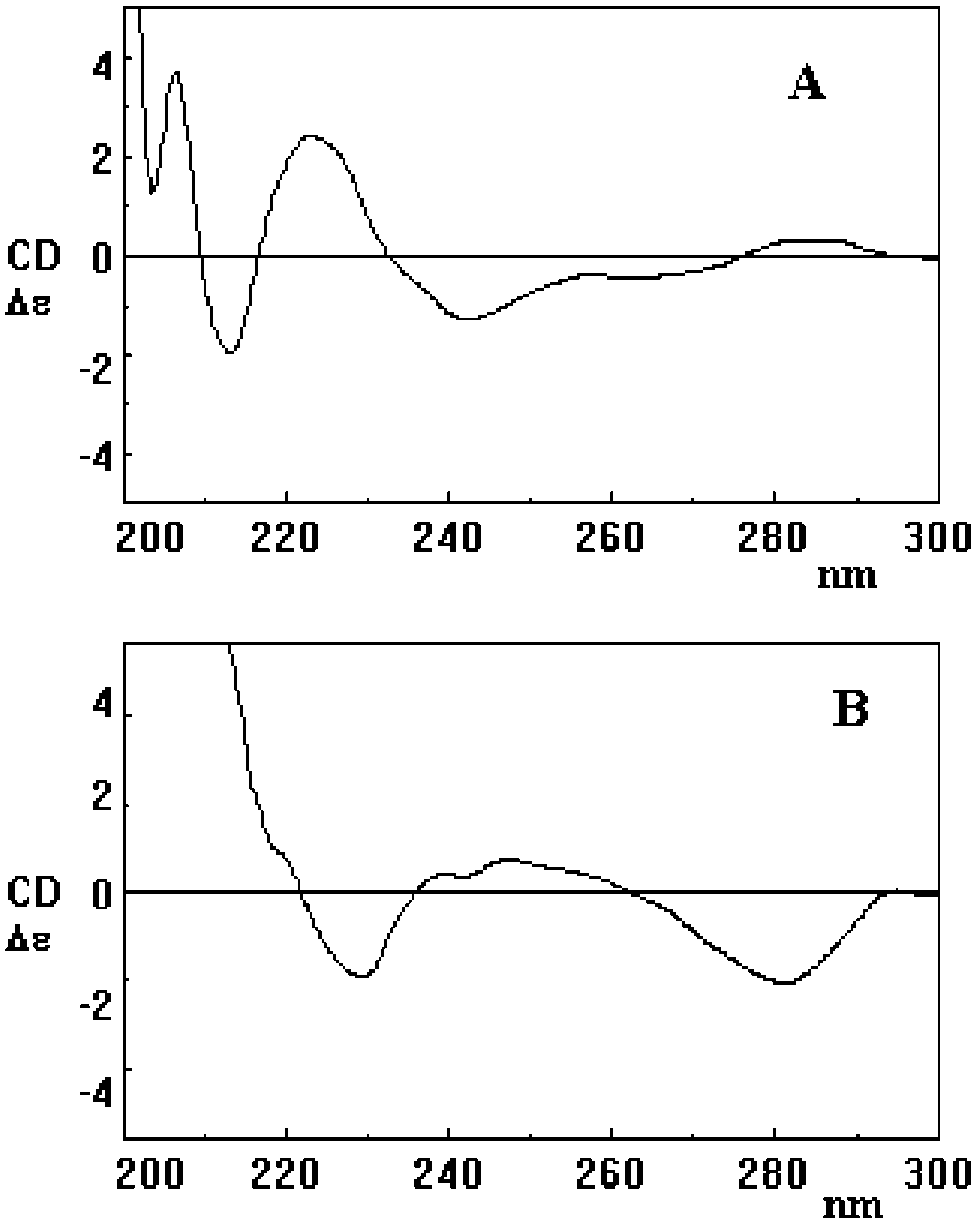 FIGURE 1. CD spectra of compounds 3a (A), 4a (B), and 7a (C).
FIGURE 1. CD spectra of compounds 3a (A), 4a (B), and 7a (C).