Tadalafil gehört zur Gruppe der PDE5-Hemmer und wirkt über eine hochselektive Blockade des Enzyms Phosphodiesterase Typ 5. Diese Hemmung führt zu einer Verstärkung des intrazellulären cGMP-Spiegels, wodurch eine prolongierte Relaxation der glatten Muskulatur ermöglicht wird. Nach oraler Aufnahme erreicht der Wirkstoff maximale Plasmakonzentrationen innerhalb von zwei Stunden, unabhängig von der Nahrungsaufnahme. Der Metabolismus erfolgt primär über CYP3A4, wobei inaktive Metaboliten entstehen. Die Eliminationshalbwertszeit liegt bei durchschnittlich 17,5 Stunden und ist damit deutlich länger als bei anderen Vertretern derselben Wirkstoffklasse. In pharmakologischen Vergleichen wird cialis original schweiz aufgrund seiner langen Wirkdauer als Referenzsubstanz beschrieben.
Cystinosis and its treatment
ARTICLES CYSTINOSIS AND ITS TREATMENT By D. Cairns, PhD, MRPharmS, R. J. Anderson, PhD, MRSC, M. Coulthard, MB ChB, and J. Terry Cystinosis is a rare inherited disease with an incidence, in developed countries, of about one case inevery 200,000 live births. In the past, it was rare for cystinosis sufferers to survive into adulthood. Thedisease occurs when the mechanism that removes excess cystine breaks down. Cystine then accumulateswithin body cells preventing these cells from functioning correctly. This initially leads to kidneyproblems and progresses to other parts of the body, including the thyroid gland, eyes and liver.Impaired growth is yet another symptom of the condition. In this article, the condition and its
Nephropathic cystinosis is a rare The disease is caused by a defect in the cystinosis, or may be unrelated to the condi-
lysosomal transport mechanism for cystine
tion. The daily drug regimen of a typical
cystinotic patient is presented in Panel 3.
This patient is post-transplant and is receiv-
115,000 and one in 179,000 live births.1,2
ing, in addition to cysteamine and carnitine,
There are approximately 200 patients in the
the cystine transport protein, cystinosin.5,6
A number of mutations have been described,
itself in raised intracellular levels of the
as well as a major deletion present in about
The main drug treatment for cystinosis is
essential amino acid cystine to 50 to 100
50 per cent of cystinosis patients of Western
administration of the aminothiol, cysteamine
times normal levels. Crystals of cystine are
European ancestry (although cystinosis has
(mercaptamine, as the bitartrate salt, Cysta-
been described in all major ethnic groups).
rates, leukocytes, cornea and conjunctiva.
The disease is characterised by poor growth,
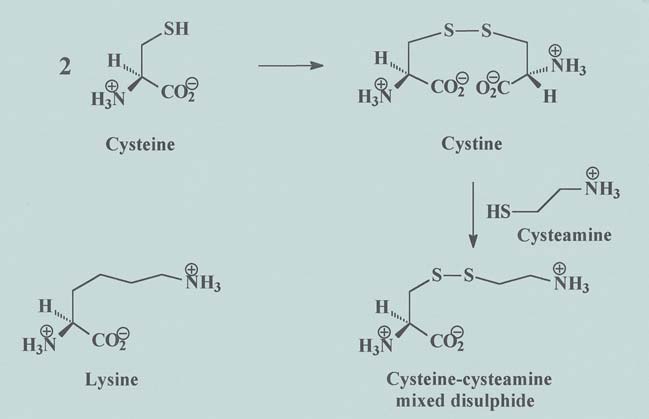
cysteamine-cysteine mixed disulphide within
URRENT TREATMENT
cells, which is structurally similar to the amino
proximal tubule function), renal glomerular
Treatment of cystinosis involves administra-
acid lysine and can egress the lysosome using
failure and involvement of other tissues and
tion of glucose and electrolytes to reverse
the pathway for lysine excretion (Figure 1).12
the effects of Fanconi syndrome, as well as
Cystinosis is fatal if not treated and death
corneal and renal transplant. Indomethacin
occurs in the second decade of life. Treat-
is administered for its sodium, potassium
molecule possesses an offensive taste and
ment begun just after birth can attenuate the
smell and irritates the gastrointestinal tract,
rate of renal failure. However glomerular
patients, carnitine is used to combat the
effects of muscle weakness brought about by
administration. In addition, cysteamine is
(approximately one year) is irreversible and
urinary loss of free carnitine and subsequent
excreted in breath and sweat, which leads to
may result in the need for renal transplant.
reduction in the transport of fatty acids into
The condition has been recently reviewed.3,4
effects, such as neutropenia. As a result of
growth in short children with chronic renal
these problems, patient compliance is poor. Panel 1. Symptoms of FUTURE TREATMENTS cystinosis
a cystinosis patient is often considerable,
especially when serious medical conditions
amine, two recent projects have been estab-
such as epilepsy or diabetes, are present.
lished at the University of Sunderland. A
into a sustained release form.13 This, it is
hoped, will minimise the gastric irritation
Panel 2. Treatments
experienced by patients taking large oral
for cystinosis Dr Cairns and Dr Anderson are senior lecturersin the Institute of Pharmacy, Chemistry andBiomedical Sciences, University of Sunderland.Dr Coulthard is a paediatric nephrologist, RoyalVictoria Infirmary, Newcastle-upon-Tyne, andMr Terry is founder and adviser, CystinosisFoundation UK. Correspondence to Dr Cairns(e-mail [email protected])ARTICLES Panel 3. Daily drug regimen for a typical patient with cystinosis
Cysteamine bitartrate 750mg tidSodium valproate
This patient is post-transplant and is receiv-
Figure 1: Structural similarity between (a) lysine and (b) cysteine-cysteamine mixed
ing, in addition to cysteamine and carnitine,
disulphide
treatment for epilepsy, diabetes andhypothyroidism
teamine. Prodrugs often exhibit desirable
improvements over the parent drug, such as
ber of cysteamine prodrugs and determined
increased lipophilicity (which aids uptake
their general cytotoxicity in cultures of expo-
cells.14 Preliminary results indicate that none
mouse model (an animal with an engineered
of the prodrugs tested showed any cell toxicity
genetic defect that results in it displaying the
USEFUL WEBSITES
up to a concentration of 100mM. These com-
symptoms of cystinosis). Study of cystinosis
pounds are currently being evaluated for their
has long suffered from the lack of a naturally
ability to deplete cystine in cultured cystinotic
cells. Such research offers hope for the future
cystinosis” will enable novel therapies to be
of cystinosis sufferers. If successful, the pro-
evaluated quickly. The most far ranging and
drug approach for cystinosis will target cyste-
potentially exciting use for these animals is
as a target for gene therapy. Once the mouse
drastically reduce side effects and eliminate
the need for repeated daily dosing. The devel-
opment of prodrugs for cystinosis would be
functioning CTNS gene. Successful genetic
greatly facilitated should a pharmaceutical
cystinotic mouse would be a necessary pre-
human patients with cystinosis (J. G.
the possibility of improved diagnosis for the
REFERENCES
Haycock GB, Al-Dahan J, Mak RHK, Chantler C. Effect of
Manz F, Gretz N. Cystinosis in the Federal Republic of Ger-
indomethacin on clinical progress and renal function in cysti-
many. Coordination and Analysis of the data. J Inher Metab
nosis. Arch Dis Child 1982;57:934–9.
Gahl WA, Bernardini I, Dalakas M, Rizzo WB, Harper GS,
Ebbensen F, Mygind KI, Holck F. Infantile nephropathic
Hoeg JM. Oral carnitine therapy in children with cystinosis
cystinosis in Denmark. Dan Med Bull 1976;23:216–22.
and renal Fanconi syndrome. J Clin Invest 1988;81:549–60.
Bois E, Feingold J, Frenay P, Briard ML. Infantile cystinosis
10. Haffner D, Wuhl E, Schaefer F, Nissel R, Tonshoff B, Mehls
in France: genetics, incidence, geographic distribution. J Med
O. Factors predictiveof the short- and long-term efficacy of
growth hormone treatment in prepubertal children with
Gahl WA, Thoene JG, Schneider JA. Cystinosis: a disorder of
chronic renal failure. J Am Soc Nephrol 1998;9:1899–907.
lysosomal membrane transport. In: Scriver CR, Beaudet AL,
11. Wilson DP, Jelley D, Stratton R, Coldwell JG. Nephropathic
Sly WS, Valle D, Vogelstein B (editors). The metabolic and
cystinosis: improved linear growth after treatment with
molecular basis of inherited disease (8th ed). New York:
recombinant human growth hormone. J Paed 1989;115:
Town M, Antignac C. Molecular characterization of CTNS
12. Pisoni R, Thoene J, Christtensen H. Detection and charac-
deletions in nephropathic cystinosis: development of a PCR-
terization of carrier mediated cationic amino acid transport in
based detection assay Am J Hum Genet 1999;65:353–9.
lysomsomes of normal and cystinotic fibroblasts. Role in
Forestier L, Jean G, Attard M, Cherqui S, Lewis C, van’t
therapeutic cystine removal? J Biol Chem 1995;260:4791–8.
Hoff W, et al. Oral carnitine therapy in children with cysti-
13. Owen B, Rowley G, Sharkey IM, Coulthard MG. Develop-
nosis and renal Fanconi syndrome. J Clin Invest 1988;
ment of cysteamine hydrochloride pellets for cystinotic
infants. Eur Hosp Pharm 1997;3:136–42.
Betend B, Pugeaut R, David L, Hermier M, Francois R. Pae-
14. Cardwell WA, Cairns D, Anderson RJ. Synthesis and evalua-
diatric cystinosis: an experience with indomethacin therapy.
tion of cysteamine analogues. J Pharm Pharmacol 1997;
• International Prostate Symptom Score over the age of 50 is an enlarged prostate. (IPSS) or AUA Symptom Index — a short this problem. By age 85, the number climbsor enlarged prostate and how often theyoccur. Due to the prostate’s location, benignenlargement may result in obstruction of • Urinalysis — a laboratory test of your urine performed to rule out the presence pro
F O R M A T O E U R O P E O P E R I L C U R R I C U L U M INFORMAZIONI PERSONALI COSTANZA GIULIANI VIA DI SCOPINO, 56 50019 SESTO FIORENTINO, FIRENZE 055/4218488 3381606286 [email protected] ESPERIENZE LAVORATIVE • Date (da – a) • Nome e indirizzo del datore di Azienda Ospedaliero-Universitaria Careggi • Tipo di azienda o set ore
 ARTICLES
ARTICLES