Tadalafil gehört zur Gruppe der PDE5-Hemmer und wirkt über eine hochselektive Blockade des Enzyms Phosphodiesterase Typ 5. Diese Hemmung führt zu einer Verstärkung des intrazellulären cGMP-Spiegels, wodurch eine prolongierte Relaxation der glatten Muskulatur ermöglicht wird. Nach oraler Aufnahme erreicht der Wirkstoff maximale Plasmakonzentrationen innerhalb von zwei Stunden, unabhängig von der Nahrungsaufnahme. Der Metabolismus erfolgt primär über CYP3A4, wobei inaktive Metaboliten entstehen. Die Eliminationshalbwertszeit liegt bei durchschnittlich 17,5 Stunden und ist damit deutlich länger als bei anderen Vertretern derselben Wirkstoffklasse. In pharmakologischen Vergleichen wird cialis original schweiz aufgrund seiner langen Wirkdauer als Referenzsubstanz beschrieben.
Mpi-halle.de
J. Phys.: Condens. Matter 9 (1997) 10739–10748. Printed in the UK Magnetism of free and supported vanadium clusters
S E Weber†, B K Rao†, P Jena†, V S Stepanyuk‡, W Hergert§,K Wildberger , R Zeller and P H Dederichs† Physics Department, Virginia Commonwealth University, Richmond, VA 23284-2000, USA
‡ Max-Planck-Institut f¨ur Mikrostrukturphysik, Weinbergweg 2, D-06120 Halle, Germany
§ Fachbereich Physik, Martin-Luther-Universit¨at, Fr.-Bach-Platz 6, D-06099 Halle, Germany
Institut f¨ur Festk¨orperforschung, Forschungszentrum J¨ulich, D-52425 J¨ulich, Germany
Abstract.
Magnetic properties of free and supported Vanadium clusters of up to four atoms
have been calculated self-consistently using the density functional theory. For the free clusterswe have used the self-consistent field–linear combination of atomic orbitals–molecular orbitaltheory with a Gaussian basis for the atom.
The geometries, together with the preferred
spin multiplicities, were optimized by using the method of steepest descent. For supportedclusters on Cu(001) and Ag(001), we have used the self-consistent Korringa–Kohn–Rostoker(KKR)-Green’s function approach. Both free and supported clusters are found to be magnetic,although the magnetic moments depend strongly on the cluster size.
have ferromagnetic ground states, the supported V clusters in general prefer antiferromagneticconfigurations. The role of inter-atomic distances, coordination, and surface morphology on themagnetic properties of V clusters are discussed. 1. Introduction
The ability of experimentalists to synthesize materials with reduced size (clusters andnanostructures) and dimension (one dimensional chains and multilayers), has given rise toa renewed interest in the study of magnetism of atomically engineered materials [1, 2]. It iscommonly believed that the magnetic moment per atom is enhanced when the system’s sizeand/or dimensionality is lowered. This is because both these factors reduce the coordinationnumber which in turn reduces the bandwidth [3]. Consequently, the density of states nearthe Fermi energy is enhanced causing the moments to rise. Experiments [2, 4] on clusters,nanostructures and ultra-thin films support this qualitative description.
The fact that reduced size and dimension can enhance magnetism of ferromagnetic
materials has led to numerous investigations (both experimental and theoretical) whichprobe possible magnetism in otherwise non-magnetic materials [5–8]. For example, it waspredicted [5, 6] and later verified experimentally [7] that alkali metal clusters as well as Rhclusters [8] could be magnetic. Theoretical calculations of 3d transition metal monolayerson metal substrates [9, 10] have also predicted substantial moments and magnetic ordering. Some of these predictions have been verified experimentally [1].
One of the most controversial results on magnetism of otherwise non-magnetic material
in reduced size and dimension has to do with vanadium.
theoretical calculations with varying degrees of approximation have predicted V monolayersto be magnetic [10, 11]. However, there are conflicting experimental results. While spin-polarized photoemission measurements provided no evidence [12] for ferromagnetism of V
on Ag(001), magnetism of ultrathin V layers sandwiched in Ag layers has been observedusing a SQUID magnetometer [13]. Recently, experimental investigation of V layers onMo substrates support the latter conclusion [14].
The study of magnetism of free V clusters also suffers from the same controversy. An
early experiment by Akoh and Tasaki [15] confirmed that small particles of vanadium inthe size range 100–1000 atoms are magnetic. However, a recent experiment of Douglasset al [16] found no evidence of magnetism in free V clusters containing as few as 9 atoms. This disagreement between two experimental results is striking, as it is well known that theproperties of clusters approach bulk behaviour as their size increases. How is it possible,then, that large V clusters are magnetic, while small clusters are not?
The disagreement among theoretical results is less severe. A number of groups have
studied the magnetism of V clusters containing up to 15 atoms [3, 17, 18]. These clusterswere confined to the bulk bcc geometry and bulk interatomic spacing. All calculationspredict that small V clusters are magnetic although the magnitudes of the magnetic momentsdiffer depending on the level of approximations used. It is, however, hard to compare theseresults with experiments on free clusters for the following reasons: (i) The geometry ofsmall clusters does not resemble the atomic arrangement in the corresponding bulk. (ii) Theinteratomic spacings in small clusters are usually less than that in the bulk. Both thesefactors are well known to have a significant effect on the magnetic moment.
Recently, magnetism of V islands on Ag(001) has been studied using the empirical
tight-binding approach [19]. The authors find the islands to be magnetic. Furthermore,the moments depend on the nature of the coupling between the V atoms and the nearestneighbour sites. No experiments are available to our knowledge to compare with the aboveprediction.
In this paper we study the electronic structure, relative stability and magnetism of
4) clusters in free space as well as those deposited on Ag(001) and Cu(001)
The surfaces of Cu and Ag both have the electronic d-bands located well
below the Fermi level. The influence of the noble metals on clusters is mainly due to thehybridization of the cluster d-electrons with the s- and p-levels of the substrate. Our aimis to see how the interaction of clusters with substrate atoms affects their electronic andmagnetic properties compared to those in free space. This understanding is important sinceefforts are currently being made in synthesizing cluster assembled materials by depositingthem on various substrates [20]. Our calculations are performed using state-of-the-art firstprinciples theoretical techniques. The study of free clusters is carried out using the self-consistent field–linear combination of atomic orbitals–molecular orbital (SCF–LCAO–MO)theory [21]. The calculations on V clusters supported on Ag and Cu surfaces are performedusing the self-consistent KKR-Green’s function method [22]. Both theories make use ofthe spin density functional formalism. For free clusters we have used both the local spindensity approximation (LSDA) as well as the generalized gradient approximation. Forsupported clusters, only the LSDA form is used. The studies are carried out for differentspin multiplicities, magnetic coupling and geometry.
In the following section we briefly outline the theoretical procedures used for the free
and supported clusters. The results are described in section 3 and summarized in section 4. 2. Theoretical procedure
The theoretical techniques used for studying free and supported clusters are different andare outlined separately in the following. Magnetism of free and supported vanadium clusters
The wavefunction ψ σ for the cluster is represented by a molecular orbital constructed from a linear combination of atomic orbitals φσ , centred at individual atomic sites Rν, given by ψ σ (r) = cσ φσ r − R
The coefficients of linear combination cσ are obtained by solving the following secular
equation (in atomic units) self-consistently:
+ Ves + V σ ψσ (r) = Eσ ψσ (r).
Here Ves and Vxc are the electrostatic and exchange-correlation potentials respectively. Theexchange-correlation potential was treated at two levels of theory: the local spin densityapproximation and the generalized gradient approximation in the density functional theory. The atomic wavefunctions φσ are fitted to a set of Gaussian functions and the total energy ofthe cluster for a fixed spin multiplicity is calculated self-consistently using the Gaussian 94software [23]. The LSDA calculations used the exchange and correlation functional dueto Vosko et al [24], while the generalized gradient approximation (GGA) made use of theBecke–Perdew–Wang method (BPW91) [25]. We have performed all electron calculationsusing a (14s, 9p, 5d/9s, 5p, 3d) basis for V. The forces at the atomic sites were calculatedusing the numerical gradient technique and the geometry is optimized using the method ofsteepest descent. The binding energy, Eb(n) of a Vn cluster is defined as
Eb = − E(n) − nE0
where E(n) is the total energy of the n-atom cluster and E0 is the energy of the atom. Thevertical ionization potential is calculated by taking the difference between the total energiesof the neutral and singly charged cluster, both having the neutral geometry.
The calculational method used for the supported clusters is described here only briefly, sincedetails can be found elsewhere [22]. The calculations are carried out using the KKR-Green’sfunction method. The method is based on density functional theory in the local spin densityapproximation (LSDA). The Green’s function of the bulk system is transformed into a layerrepresentation. The atomic potentials of seven layers are removed to create two practicallyuncoupled half-crystals. The Green’s function of the ideal surface is then used as thereference Green’s function to calculate the electronic properties of supported clusters. Thecluster on the surface destroys 2d translational symmetry of the ideal surface. Therefore theGreen’s function of the cluster is calculated from a Dyson equation in site representation:
Gnn (E) = G0nn (E) +
Here Gnn (E) is the energy-dependent structural Green’s function matrix and G0nn (E) the
corresponding matrix for the ideal surface. The summation is over all lattice sites n andall angular momenta L for which the perturbation
t n (E) between the t matrices of the
real and the reference system is significant. In our calculations perturbations on the nearestneighbour sites of the supported cluster are taken into account. While angular moments upto l = 3 are included in the calculations of the wavefunction, the Coulomb and exchange-correlation potentials were calculated using the full charge density by taking into account amultipole expansion up to l = 6. For the latter the functional of Vosko et al [24] is used.
Only the spherical symmetric part of the potential inside the Wigner–Seitz sphere is usedto calculate the Green’s function. This approximation is not important for the calculationof local moments [26]. 3. Results and discussion
The equlibrium geometry, binding energy and preferred spin multiplicities of free Vn (n
clusters were calculated using the SCF–LCAO–MO method. To test the accuracy of themethod we first compare our calculated results on the ground state of the V atom, as well asthe energetics of the V2 dimer, with the corresponding experimental results [27]. The groundstate of the V atom in the local spin density approximation is 6D1/2. This corresponds to amagnetic moment of 5 µB . However, when corrections to the exchange-correlation potentialare applied, based on Becke’s generalized gradient approximation (GGA), the ground stateof the V atom using the above basis set is 4F3/2 with a moment of 3 µB in agreement withexperiment [27]. We have calculated the free V atom also by means of our KKR technique. If we place a single V atom in the middle of the vacuum layers, which cuts the bulk Agsystem into two Ag(001) half-crystals, we get a magnetic moment of 4.68 µB in the Wigner–Seitz cell. This is in agreement with the LSDA version of the SCF–LCAO–MO calculation. We recall that in the KKR formalism the exchange-correlation potential is treated withinthe LSDA and non-integer occupancies are allowed. The small difference between 5.0 µBcalculated for the free atom within the SCF–LCAO–MO scheme and 4.68 µB in the KKRscheme is partly due to the small influence of the Ag substrate in the latter method andpartly due to the non-integer occupancies allowed in these calculations. Table 1. Total energies, binding energies per atom, bond lengths and magnetic moments per atom of free Vn clusters using the LSDA and non-local correction (GGA).
The magnetic moments of the V adatom (see table 2) on the Cu(001) and Ag(001)
substrates are respectively 3.03 µB and 3.41 µB and are considerably reduced from the freeatom value of 4.68 µB . This is caused by the hybridization of the V d-electrons with thesp-electrons of the metal substrates and by the nearly total quenching of the s-moment. Thishybridization is stronger for the Cu(001) substrate than for the Ag(001) substrate.
The calculated ionization potential of the V atom is 6.12 eV and compares well with the
experimental value [27] of 6.74 eV. The electronic ground state of V2 is 3
with recent experiment [28]. The calculated binding energy and bond length of V2 at theGGA level of theory are respectively 2.94 eV and 1.73 ˚
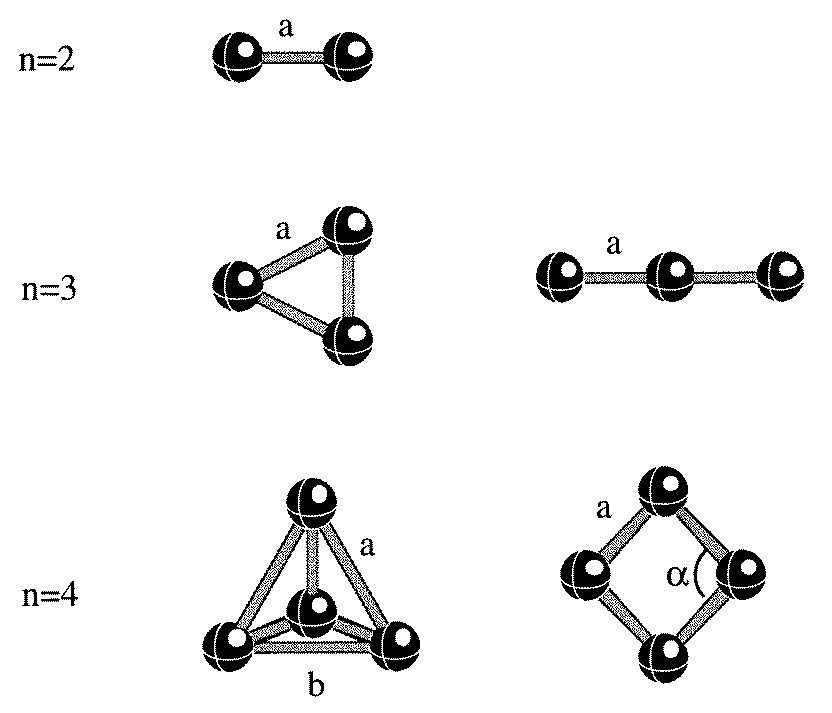
Magnetism of free and supported vanadium clustersFigure 1. Equilibrium geometries of free Vn clusters. The geometrical parameters are given in table 1.
A. We should note that LSDA tends to overestimate the
binding energy while the bond length is less sensitive. For example, we obtain a bindingenergy of 5.52 eV and a bond length of 1.71 ˚
give the ground state of V2 to be a triplet. There have been a number of earlier theoreticalworks [29] on V2 ranging from extended H¨uckel to local spin density calculations, whichyield the ground state of V2 to be 9
− or 1 +. However, our result of spin triplet as the
ground state of V2 agrees with recent experiment [28] and theory [3, 30].
The geometries of the Vn clusters are given in figure 1. The bond lengths, binding
energies, and the magnetic moments per atom are given in table 1. Note that, as in V2, thebinding energy per atom in the local spin density approximation is consistently higher thanthat using the gradient correction. However, the bond lengths, bond angles and the magneticmoment per atom remain practically unaffected. It is also interesting to note that the bindingenergy of 1.47 eV/atom and bond length of 1.73 ˚
than the corresponding values for bulk vanadium, which has a cohesive energy of 5.31 eVand a nearest neighbour distance of 2.62 ˚
A. The magnetic moment per atom of the dimer is
1.0 µB and is significantly reduced from the gradient corrected free atom value of 3.0 µB .
To aid in the discussion to follow, we have calculated the binding energy of a V2 dimer
as a function of interatomic distance for different spin multiplicities, such as triplet, quintet,septet, etc. It is expected that as the interatomic separation of the V2 dimer is increased,the overlap between the orbitals centred at each atomic site should decrease. This in turnshould cause the magnetic moment per atom to increase. To examine how this processevolves, we plot the binding energy (calculated using the GGA) of the dimer in figure 2 forvarious spin multiplicities. Note that when the interatomic separation reaches about 1.9 ˚
the ground state changes from a triplet to quintet. At an interatomic distance of about 2.2 ˚
the states with spin multiplicities of 5 and 7 are nearly degenerate. Beyond this distance thepreferred spin multiplicity is 7. Thus, as V clusters are deposited on a metallic substrate,their interaction with the substrate atoms will cause the V–V distance to conform to thedistance in the underlying lattice. Consequently, this factor alone can cause their magneticmoment to differ from the free cluster value.
For example, when we confine the V2 dimer to a distance of 2.56 ˚
Figure 2. Binding energies Eb for the V2 dimer as a function of interatomic distance and spin multiplicities. Note that the energies are based on the generalized gradient approximation in the density functional theory. Table 2. Magnetic moments of ferromagnetic states of Vn clusters on Cu(001) and Ag(001) surfaces (Cn—chains of n atoms, I4—island of 4 atoms). For the trimer C3, having two non- equivalent sites, only the average moment per atom is given.
which corresponds to distances the dimers would have on a Cu(001) and Ag(001) surface,respectively, the magnetic moment per atom is 3.0 µB . Using the KKR formalism we havecalculated the magnetic moment per atom of V2 on the Cu(001) and Ag(001) surfaces aswell as in the bulk systems. In order to check the accuracy of the method, we have alsoused the KKR method to simulate the free V2 dimer as was done for the free V atom in thediscussion above. Our calculated values of 3.45 µB /atom for the Cu(001) and 3.58 µB /atomfor the Ag(001) surfaces agree well with the spin septet configuration obtained in the SCF–LCAO–MO method (see table 2). Note again that the KKR method only used the LSDAlevel of theory while our results in figure 2 are with generalized gradient approximation. However, the use of the LSDA (as opposed to the GGA) method in the KKR calculation isnot expected to adversely affect our conclusions concerning the magnetic moments of thesupported V clusters. As mentioned earlier and shown in table 1, the magnetic momentsare not sensitive to these approximations in clusters.
The interaction of the dimer with the Ag surface reduces the moment to 3.38 µB per
Magnetism of free and supported vanadium clusters
atom. This is mostly due to the further quenching of the s-moments on the surfaces. Thesmaller interatomic spacing on the Cu(001) surface compared to that of Ag and a strongerhybridization of the V wavefunctions with the sp-wavefunctions of the Cu substrate reducesthe moment of the supported V2 dimer further to 2.85 µB . Bl¨ugel et al [10] have alsocalculated the moments of single V impurities and V impurity pairs in bulk Cu and Ag. In this paper it was shown that the strength of the 3d–3d hybridization increases with thedelocalization of the 3d wavefunction, due to the reduction of the dimer bond length. Ourresults on the relative variation of magnetic moments of V2 dimers on Cu and Ag areconsistent with this study.
The structure of the free V3 trimer is an equilateral triangle with a bond length of 2.14 ˚
and a binding energy of 1.76 eV/atom. The magnetic moment per atom is 1.67 µB andis significantly higher than the dimer value of 1.0 µB . To understand the origin of thisincrease, we note that the interatomic distance as well as the coordination number in V3 arelarger than those in V2. Typically these two factors act in opposite directions—increasingthe interatomic distance causes the moments to rise (as seen in figure 2), while increasing thecoordination number causes the moments to decrease. If V2 is constrained to a bond lengthof 2.14 ˚
A, the preferred moment per atom for this configuration would be 2 µB /atom. The
fact that in V3 the magnetic moment is 1.67 µB /atom shows that the increasing coordinationhas played a role in diminishing the magnetic moment.
Brune et al [2] have shown that small linear chains and planar islands can be created on a
substrate by special growth techniques. Therefore, we have studied the magnetic momentsof a V3 linear chain and V4 in-plane square island configuration. Other geometries arepossible, but here we only restrict ourselves to the above geometries.
The moments of the supported trimers are changed only slightly (2.76 µB for Cu(001)
and 3.36 µB for Ag(001)) compared with the dimers. The atom in the middle of the trimerhas a higher coordination and therefore a smaller moment (2.58 µB for Cu(001) and 3.34 µBfor Ag(001)) than the other two atoms (2.85 µB for Cu(001) and 3.38 µB for Ag(001)). This leads to a decrease of the mean value of the trimer moment (see table 2).
We have also calculated the moment of the V3 trimer as a linear chain in free space. We
find that there are two magnetic solutions with 2.33 µB /atom and 1.0 µB /atom that are nearlydegenerate, the former being approximately 0.01 eV lower in energy. This configuration,however, is 1.5 eV higher in energy than the groundstate, which is an equilateral triangle.
For a V4 cluster, the equilibrium structure is a slightly distorted tetrahedron. The
interatomic distance in the plane is 2.33 ˚
A while that between the apex atom and an atom
A. The binding energy per atom and the magnetic moment per atom are
respectively 2.20 eV and 1.0 µB . The sharp decrease in the moment in V4 compared to thatin V3 can be attributed to be primarily due to an increasing coordination number since theinteratomic distances between V3 and V4 clusters differ very little. We have also searchedfor the existence of other structural isomers and found that V4 can exist as a planar rhombusstructure with an interatomic distance of 2.06 ˚
A. However, this structure lies 0.21 eV in
energy above the tetrahedral configuration. Interestingly, both the rhombus and tetrahedralstructure have the same moment per atom at the GGA level of theory.
For the supported islands consisting of 4 atoms the moments decrease compared to the
trimers. The decrease for the Cu substrate is similar to what has been observed for the freeclusters. For the four-atom cluster on the Ag substrate the relative decrease is considerablysmaller. The decrease of the moment on Cu is stronger than that on Ag, since the latticeconstant of Cu is about 10% smaller than that of Ag and therefore the d–d hybridization isstronger.
It should be noted that in the supported clusters, the effect of relaxation was ignored. For
example, the atomic positions in Cu(001) and Ag(001) substrates were assigned their ideallattice configurations. When V clusters were deposited on these substrates, their positionswere also assumed to conform to the ideal substrate structure, i.e. the V–V distance andheight of the cluster plane from the surface were the same as that of the substrate. Thisassumption was necessary due to the computational limitations. The loss of symmetrydue to the absorbed clusters already makes the ab initio KKR calculations very complex. Adding the effect of surface relaxation is beyond the scope of present day computationalresources if calculations at the first principles level of theory, taking into account the semi-infinite surface, are desired. However, we can study the effect of relaxation qualitativelyby referring to our results in figure 2. Note that the nearest neighbour distances in bulkV, Cu, and Ag are respectively 2.62, 2.56 and 2.89 ˚
not expect any significant change in the moment due to relaxation. For Vn on Ag(001) thelattice mismatch is about 10%. As can be seen from figure 2, the moment per atom of theV2 dimer does not change for 2.6 < R < 2.9 ˚
A. Consequently we expect the moment of Vn
clusters on Ag to be unaffected due to relaxation as well. This expectation, of course, clearlyneglects any electronic considerations arising from the interaction between the clusters andthe substrates. We are currently working on methods that combine KKR with the tightbinding model, which will enable us to examine, in the future, the role of surface relaxationon cluster magnetism.
In this paper we have restricted our studies of Vn clusters to the ferromagnetic solutions.
This is because the Vn clusters in free space are found to be ferromagnetic. However, thisseems not to be the case for the supported V clusters. Detailed calculations for Cu(001) [33]show that the V dimers, trimers and tetramers prefer antiferromagnetic configurations. Forinstance, the antiferromagnetic configuration of the V2 dimer is 0.2 eV/atom lower in energythan the ferromagnetic one, and also for the V trimer and tetramer the configurations withantiferromagnetic nearest-neighbour coupling are lower in energy than the ferromagneticsolutions, in the case of V3 by 0.11 eV/atom and V4 by 0.05 eV/atom. Since all freeV clusters are ferromagnetic, we believe that the preference of the antiferromagneticconfiguration is a typical result of the hybridization with the substrate.
we have calculated the ferromagnetic and antiferromagnetic state of the free V dimer witha bond length corresponding to the nearest neighbour distance of the Ag lattice using theKKR method. We get moments of 3.58 µB for the ferromagnetic state and ±3.58 µBfor the antiferromagnetic state. The ferromagnetic groundstate is 0.013 eV/atom lower inenergy than the antiferromagnetic state. This is in line with calculations by Bl¨ugel et al[10] for complete V monolayers. While a V monolayer deposited on Ag(001) prefers theantiferromagnetic configuration, a free-standing V monolayer with the Ag lattice constantshows a ferromagnetic ground state. As explained in detail by Bl¨ugel et al
hybridization with the substrate slightly broadens the density of states of the monolayers,which is unfavourable for the ferromagnetic configuration.
Another characteristic property of the multimers on surfaces is the existence of multiple
spin configurations. This effect has been known to exist for bulk metals, alloys, as wellas ultrathin films [31].
Even some clusters exhibit multiple spin configurations.
example, the anomalous photo-detachment spectra of the Li− cluster was explained due
to the coexistence of the anionic cluster in doublet and quartet spin configurations [32]. Recently Stepanyuk et al [33] have studied the energetics of the trimers of 3d elements onCu(001). For all 3d trimers they found the existence of four different spin configurations:ferromagnetic, antiferromagnetic and two different kind of low spin states.
particular cases such as V3 and Mn3, the energy differences between various magneticstates can be as small as 8 meV and magnetic fluctuations are possible. In general also
Magnetism of free and supported vanadium clusters
non-collinear states cannot be excluded.
The effect of the matrix on the magnetic properties of the transition metal cluster will
undoubtedly be an important topic for further studies, as the possibility of synthesizingcluster assembled materials becomes promising. Thus an in depth understanding of theproperties of the free and supported clusters will lead to a rich and interesting field ofresearch in the future. 4. Conclusions
Self-consistent calculations based on the local spin density functional theory have beencarried out to study the magnetic properties of free and supported V clusters on Cu(001)and Ag(001) substrates. The geometries of the free clusters were fully optimized. All freeVn clusters up to n = 4 were found to be magnetic with moments per atom varying non-monotonically with size. While the non-local correction to the density functional theoryhad a substantial effect on the magnetic moment of the free V atom, it had no effect onthe moment of V clusters when compared to the local approximation. We recall that Vnclusters containing more than nine atoms were found to be magnetic experimentally. In thiswork we have not optimized geometries of Vn clusters for n > 4. Thus it is difficult tocompare the theory with experiment. However, we are confident about our results that Vn(n
4) clusters are magnetic. This confidence is based on the past success of the density
functional theory not only predicting [6] magnetism of clusters that later were verifiedexperimentally [8], but also in correctly identifying the ground state spin multiplicity of Fe,Co, and Ni dimers [34].
Surprisingly the moments of the supported V clusters are considerably larger than the
ones for the free clusters. This enhancement of the moments of the supported clustersis basically due to the fact that the nearest neighbour distances offered by the Ag(001)and Cu(001) substrates are significantly larger than the corresponding distances in the freeclusters. The somewhat smaller moments on the Cu substrate as compared to the Ag canalso be understood in this way since the lattice constant of Ag is 10% larger than that of Cu,resulting in a much reduced hybridization. Accurate description of the substrate relaxationresulting from cluster deposition is necessary before one can conclusively state that thesupported Vn clusters would retain their magnetic character.
A second difference between the examined free and supported V clusters is the magnetic
character of the ground state. While the free clusters have ferromagnetic ground states, thesupported V clusters in general prefer antiferromagnetic configurations. The hybridizationwith the substrate leads to a broadening of the density of states, which tends to destabilize theferromagnetic coupling. Our calculations also suggest the existence of multiple magneticsolutions. In special cases this might make it difficult to compare theory with existingexperiment. Thus magnetic experiments on controlled substrates with well defined clusterswould be very helpful. Acknowledgments
This work is supported in part by the Army Research office. We than S Bl¨ugel and JKirschner for helpful discussions. The computations were performed partially on Craycomputers of the Forschungszentrum J¨ulich and the German supercomputer centre (HLRZ). One of the authors (PJ) is grateful to Professor K V Rao for suggesting this problem. References
[1] Heinrich B and Cochran J F 1993 Adv. Phys. 42 523 [2] Brune H, R¨oder H, Boragno C and Kern K 1994 Phys. Rev. Lett. 73 1955
R¨oder H, Hahn E, Brune H, Bucher J P and Kern K 1993 Nature 366 141
[3] Liu F, Khanna S N and Jena P 1991 Phys. Rev. B 43 8179 [4] Apsel S E, Emmert J W, Deng J and Bloomfield L A 1996 Phys. Rev. Lett. 76 1441
Billas I M L, Chatelain A and de Heer W A 1994 Science 265 1682 Shi J, Gider S, Babcock K and Awschalom D D 1996 Science 271 937
[5] Rao B K, Jena P and Manninen M 1985 Phys. Rev. B 32 477 [6] Reddy B V, Khanna S N and Dunlap B I 1993 Phys. Rev. Lett. 70 3323 [7] Nozue Y, Kodaira T and Goto T 1992 Phys. Rev. Lett. 68 3789 [8] Cox A J, Louderback J G and Bloomfield L A 1993 Phys. Rev. Lett. 71 923 [9] Ohnishi S, Freeman A J and Weinert M 1983 Phys. Rev. B 28 6741
[10] Bl¨ugel S, Drittler B, Zeller R and Dederichs P H 1989 J. Appl. Phys. A 49 547 [11] Fu C L, Freeman A J and Oguchi T 1985 Phys. Rev. Lett. 54 2700
Gay J G and Richter R 1985 Phys. Rev. Lett. 56 2728 Mokrani A, Dreyss´e H, Bouarab S and Demangeat C 1992 J. Magn. Magn. Mater. 113 201
[12] Stampanoni M, Vaterlaus A, Pescia D, Aeschlimann M, Meier F, D¨urr W and Bl¨ugel S 1988 Phys. Rev. B
[13] Shintaku K, Mizutani T, Hosoito N and Shinjo T 1991 J. Phys. Soc. Japan 60 1078 [14] Korenivski V, Rao K V, Birch J and Sundgren J E 1995 J. Magn. Magn. Mater. 140–144 549 [15] Akoh H and Tasaki A 1978 J. Appl. Phys. 49 1410 [16] Douglass D C, Boucher J P and Bloomfield L A 1992 Phys. Rev. B 45 6341 [17] Salahub D R and Messmer R P 1981 Surf. Sci. 106 415 [18] Lee K and Callaway J 1993 Phys. Rev. B 49 15 358
Lee K and Callaway J 1994 Phys. Rev. B 49 13 906
[19] Nait-Laziz H, Demangeat C and Mokrani A 1993 J. Magn. Magn. Mater. 121 123 [20] Dupuis V, Perez J P, Tuaillon J, Pillard V, Mellinon P, Perez A, Barbara B, Thomas L, Fayeulle S and
Gay J M 1994 J. Appl. Phys. 76 6676
[21] Here W J, Random L, Schleyer P V R and Pople J A 1986 Ab initio Molecular Orbital Theory (New York:
[22] Zeller R, Lang P, Drittler B and Dederichs P H 1992 MRS Symp. Proc. 253 (Pittsburgh, PA: Materials
Stepanyuk V S, Lang P, Wildberger K, Zeller R and Dederichs P H 1994 Surf. Rev. Lett. 1 477 Lang P, Stepanyuk V S, Wildberger K, Zeller R and Dederichs P H 1994 Solid State Commun. 92 755
[23] Frisch M J 1995 Gaussian 94 Revision B.1 Gaussian Inc., Pittsburgh, PA, USA [24] Vosko S H, Wilk L and Nusair M 1980 Can. J. Phys. 58 1200 [25] Becke A D 1988 Phys. Rev. A 38 3098
Perdew J P and Wang Y 1992 Phys. Rev. B 45 13 244
[26] Wildberger K, Stepanyuk V S, Lang P, Zeller R and Dederichs P H 1995 Phys. Rev. Lett. 75 509
Stepanyuk V S, Hergert W, Wildberger K, Zeller R and Dederichs P H 1996 Phys. Rev. B 53 2121
[27] Moore C E 1970 Ionization Potentials and Ionization Limits Derived from the Analyses of Optical Spectra
NSRDS-NBS34 (Washington DC: National Bureau of Standards)
Morse M D 1986 Chem. Rev. 86 1049
[28] Spain E M and Morse M D 1992 J. Phys. Chem. 96 2477 [29] Cooper W F, Clarke G A and Hare C R 1972 J. Phys. Chem. 76 2268
Ford T A, Huber K, Klotzb¨ucher W, K¨undig E P, Moskovits M and Ozin G A 1977 J. Chem. Phys. 66 524 Harris J and Jones R O 1979 J. Chem. Phys. 70 830
[30] Salahub D R 1987 Ab initio Methods in Quantum Chemistry II ed K P Lawley (New York: Wiley) p 447
[31] Wassermann E F 1991 J. Magn. Magn. Mater. 100 346 [32] Rao B K, Jena P and Ray A K 1996 Phys. Rev. Lett. 76 2878 [33] Stepanyuk V S, Hergert W, Wildberger K, Zeller R and Dederichs P H 1997 submitted [34] Castro M, Jamorski C and Salahub D R 1997 Chem. Phys. Lett. 271 133
THE STAGES OF CHANGE APPROACH WORKPLACE SEVENTH DAY ADVENTIST CHURCH QUIT SMOKING WORKSHOP “BUTT OUT” program for military members, their CANADIAN CANCER SOCIETY’S 1-866-527-7383 The Stages of Change Approach is a workshop to help Breathe Free, 8 two-hour sessions over 3 weeks. Follow-families, and members of the defence team. SMOKERS’ HELPLINE support employees w
Lynda LAMOUDI ETUDES ET DIPLOMES 2005 : Thèse de Doctorat en cours de réalisation, intitulé du sujet : Optimisation comparative procédés d’encapsulation, l’un par granulation et l’autre par sphéronisation Université des Sciences et de la Technologie (USTHB). Inscription en codirection avec le Professeur CHAUMEIL de l’Université René Descartes. Paris5 ; 2005 :
 Magnetism of free and supported vanadium clusters
Figure 1. Equilibrium geometries of free Vn clusters. The geometrical parameters are given in
Magnetism of free and supported vanadium clusters
Figure 1. Equilibrium geometries of free Vn clusters. The geometrical parameters are given in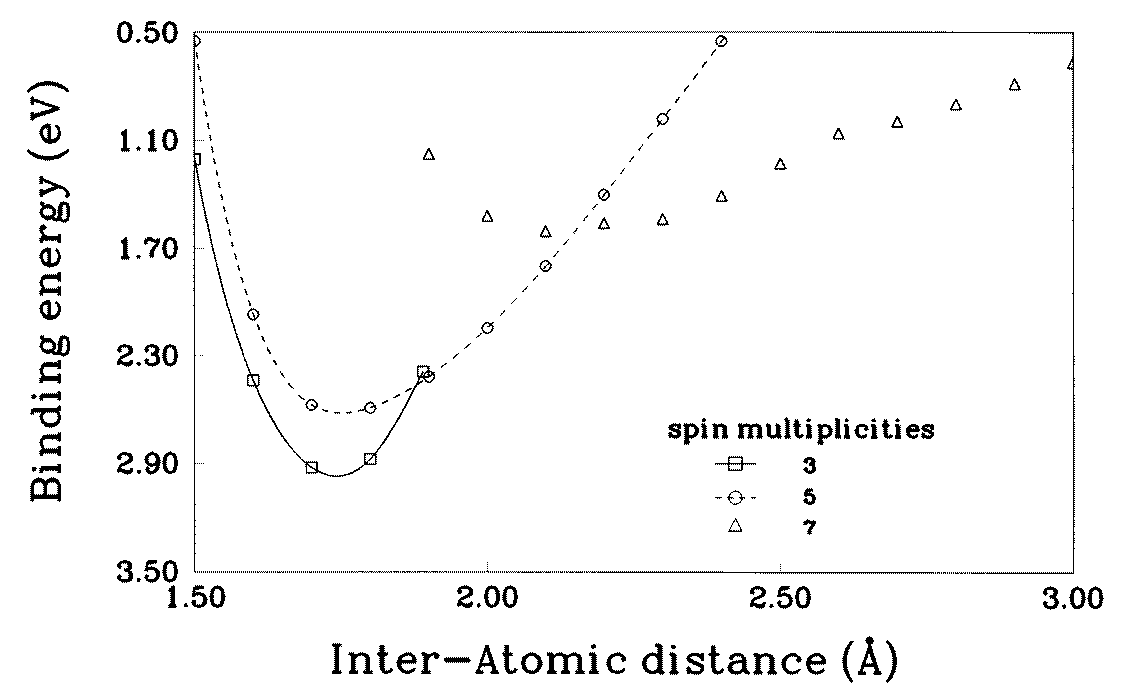 Figure 2. Binding energies Eb for the V2 dimer as a function of interatomic distance and spin
Figure 2. Binding energies Eb for the V2 dimer as a function of interatomic distance and spin